
Abstract
Congenital disorders of glycosylation (CDG) are a large group of recessive multisystem disorders caused by impaired protein or lipid glycosylation. The CDG-I subgroup is characterized by protein N-glycosylation defects originating in the endoplasmic reticulum. The genetic defect is known for 17 different CDG-I subtypes. Patients in the few reported DPAGT1-CDG families exhibit severe intellectual disability (ID), epilepsy, microcephaly, severe hypotonia, facial dysmorphism and structural brain anomalies. In this study, we report a non-consanguineous family with two affected adults presenting with a relatively mild phenotype consisting of moderate ID, epilepsy, hypotonia, aggressive behavior and balance problems. Exome sequencing revealed a compound heterozygous missense mutation, c.85A>T (p.I29F) and c.503T>C (p.L168P), in the DPAGT1 gene. The affected amino acids are located in the first and fifth transmembrane domains of the protein. Isoelectric focusing and high-resolution mass spectrometry analyses of serum transferrin revealed glycosylation profiles that are consistent with a CDG-I defect. Our results show that the clinical spectrum of DPAGT1-CDG is much broader than appreciated so far.
Similar content being viewed by others


Introduction
Congenital disorders of glycosylation (CDG) are a large class of neurometabolic disorders that involve defects in protein N- and O-glycosylation and lipid glycosylation. Defects in N-glycosylation are traditionally divided in CDG type I (CDG-I) and CDG type II (CDG-II).1 These subtypes are owing to assembly defects in the endoplasmic reticulum and to processing defects in the golgi apparatus, respectively. More than 50 CDGs are reported,2 including 17 types of CDG-I.3 One of the less-frequent types is DPAGT1-CDG, (formerly CDG-1j, MIM # 608093), which is caused by mutations in the DPAGT1 gene.4 DPAGT1 encodes UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase, the enzyme that catalyzes the first step in the dolichol-linked oligosaccharide pathway.5 So far, there are four reports in which the cause and the features of DPAGT1-CDG have been described. In 2003, Wu et al4 described the first patient with DPAGT1-CDG having a missense mutation, c.660A>G (p.Y170C) in combination with an unknown splicing defect. The patient presented with severe hypotonia, seizures, intellectual disability (ID), microcephaly and exotropia. Subsequently, Vuillaumier–Barrot reported two siblings who were compound heterozygous for a missense mutation, c.890A>T (p.I297F) and a splice site mutation, c.162-8G>A. The clinical presentation was not described.6 In 2011, Imtiaz et al7 described a third report belonging to a large Saudi Arabian family with homozygous missense mutation, c.902G>A (p.R301H). The patients presented with severe hypotonia, ID, seizures and microcephaly. The patients died in the early age. Recently, a fourth report has been illustrated by Wurde et al8, with a homozygous missense mutation, c.341C>G (p.A114G) in a consanguineous Turkish family. The patients presented with hyper excitability, seizures, bilateral cataract, progressive microcephaly and hypotonia. The patients died in early infancy.8 In this study, we report a non-consanguineous Pakistani family with two adult members affected showing a relatively mild phenotype consisting of moderate ID, epilepsy, hypotonia and speech problems. This family was classified as an ID disorder of unknown etiology. However, exome sequencing revealed a compound heterozygous mutation in DPAGT1. Transferrin isoelectric focusing (TfIEF) and high-resolution mass spectrometry supported the diagnosis of a CDG-I.
Materials and methods
Patient material
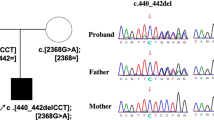
A non-consanguineous family (PKMR13) with moderate ID was ascertained from the central part of the Punjab province, Pakistan (Figure 1a). Informed consents were obtained from all the participating subjects. This study was approved by the Institutional Review Board (IRB) of the Center of Excellence in Molecular Biology (CEMB), University of the Punjab, Lahore, Pakistan and the local ethics committee of the Radboud University Nijmegen Medical Center, Nijmegen, The Netherlands. DNA isolation was carried out by using the standard protocols.9

Family structure and photographs of the patients. (a) Pedigree of PKMR13. Filled symbols indicate the affected individuals and single horizontal line indicates the marriage connection. The diagonal line indicates deceased individual. (b) Photographs of two affected (III:3 and III:4) individuals presenting with moderate ID, epilepsy, hypotonia, speech problems, aggressiveness and atypical facial dysmorphism.
Genotyping and copy number variation analysis
We have used two SNP microarray platforms (Illumina 6 k array and Affymetrix 250 k array, San Diego, CA, USA) for genotyping. We used three individuals (II:1, III:2 and III:3) to genotype on an Illumina 6 k SNP array platform. To test for copy number variations (CNVs) and for possible homozygous regions, the DNA of one affected individual (III:4) was used to genotype by Affymetrix 250 k SNP array. The Affymetrix Genotyping Console (version 2.0) and Illumina BeadStudio genotyping module software were used to generate the genotype calls. Given the high rate of consanguineous unions in Pakistan, we searched for regions of homozygosity of at least 1 Mb by visual inspection. To investigate CNVs, the Affymetrix 250 k SNP array data were analyzed by using Copy Number Analyzer for GeneChip (CNAG).10
Next-generation sequencing
DNA of patient III:4 was used for exome sequencing. Next-generation sequencing and analysis was carried out, as described before.11 In brief, exome enrichment was performed using the SureSelect Human All Exon 50 Mb Kit (Agilent Technologies, Santa Clara, CA, USA), covering ∼21 000 genes. The enriched exome library was equimolarly pooled in a set of four samples, including three other unrelated samples. The pool was on the basis of a combined library concentration of 1 pM. Subsequently, the obtained pool was used for emulsion PCR and bead preparation, using the EZbead system, following the manufacturer's instructions (Life Technologies, Carlsbad, CA, USA). For each pool of four exome libraries, a full sequencing slide was used on a SOLiD 4 System, thereby anticipating that all four samples would be represented as a quarter of the total beads sequenced (Life Technologies).
We obtained 5.34 Gb of sequence that could be mapped to the human hg19 reference genome using Bioscope software version 1.3 (Life Technologies). This resulted in a median coverage of 63-fold, with 87% of targets covered at least 10-fold. A total of 12 590 variants were detected in the coding regions and canonical splice sites. We then applied a prioritization scheme similar, as described earlier, in order to identify the pathogenic mutation.12 To this end, we excluded variants known in dbSNPv132, as well as variants from our in-house database. The latter consisted of 369 exome samples from healthy individuals and patients with other rare diseases. This analysis reduced the number of candidate DNA variants to 410.
On the basis of expectation of a recessive inheritance model, we further prioritized these variants and identified 49 candidate genes; 31 with potential homozygous variants (determined by >80% variant reads), and 18 genes with compound heterozygous variants (determined by >20% variant reads). Variants predicting synonymous changes were filtered out. The remaining variants were evaluated by inspecting the raw read alignment files (Binary Sequence Alignment/Map format (BAM files)) to verify the presence and the minimal percentage (20%) of the variant reads. All these filtering steps revealed eight variants, including four homozygous variants, as well as four compound heterozygous variants.
Sanger sequencing
All the potential variants were analyzed by Sanger sequencing. Primers for the amplification of exons carrying a DNA variant were designed by using the Primer3 program (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi).13 The primers were designed to amplify the 300–650-bp DNA segments encompassing the respective mutations. PCR amplification was carried out on 40 ng of genomic DNA with Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA). PCR amplicons were purified by using NucleoFast 96 PCR plates (Clontech Lab, Mountain View, CA, USA), according to the manufacturer’s protocol. Sequence analysis was performed with the ABI PRISM Big Dye Terminator Cycle Sequencing V3.1 Ready Reaction Kit and the ABI PRISM 3730 DNA Analyzer (Applera Corp, Foster City, CA, USA).
Biochemical assays for analysis of transferrin N-glycosylation
TfIEF in serum of patient III:3 was carried out, as described.14 For high-resolution mass spectrometry of transferrin, a 5 μl serum sample was incubated with anti-transferrin beads, as described.15 The eluate was analyzed on a microfluidic 6540 LC-chip-QTOF instrument (Agilent Technologies) using a C8 protein chip. Data analysis was performed using Agilent Mass Hunter Qualitative Analysis Software B.04.00. The Agilent BioConfirm Software was used to deconvolute the charge distribution raw data to reconstructed mass data.
Results
Family PKMR13 was ascertained as an autosomal recessive ID family. There were two affected adults (III:3 and III:4) in this family, who are currently 34 and 32 years of age (Figure 1a). Both affected individuals were normal at birth, following an uneventful pregnancy and delivery. They started saying first word at the age of 1.5–2 years. They started walking at the age of 1.5 year. Affected individuals III:3 and III:4 showed normal development and behavior till the age of 5 and 2 years, respectively. Individual III:3 developed epileptic seizures and showed aggressive behavior and hypotonia at the age of 5 years. Patient III:4 developed epileptic seizures, aggressive behavior and hypotonia at the age of 2 and a half years, 6 months after recovery from measles and typhoid. This infection appears to be irrelevant for the phenotype of patient III.4, given the fact that an identical phenotype was observed in patient III.3, who never had measles or typhoid. Abnormal behavior was also noted by the physician, who diagnosed mental illness for both affected members and prescribed medication for epilepsy. No records of previous medication or investigations were found at the time of sample collection in 2008. Presently, the affected siblings show mild atypical facial dysmorphism (Figure 1b) aggressive behavior, hypotonia and epilepsy. They also have very poor speech and presented a moderate ID. Head circumference of both patients was normal. Additionally, patient III:3 had night blindness (Table 1). The phenotypic features of affected individuals are not progressive.
Genome wide genotyping and CNV analysis was performed by using two different SNP array platforms (6 k Illumina and 250 k Affymetrix). Genotyping by using Illumina 6 k SNP array analysis was carried to perform homozygosity mapping. Whereas, Affymetrix 250 k SNP array was performed to allow the detection of CNVs. As the resolution of Illumina 6 k array was not high enough to use it for CNV calling. No pathogenic CNVs were detected in this family. SNP genotyping revealed two shared small homozygous regions on chromosomes 19p12 (1.7 Mb) and 21q21.3 (4.1 Mb). No massive homozygous regions were detected, which is in-line with the fact that parents were not related.
To investigate the genetic cause in this family we conducted an exome sequencing approach. The exome of patient III:4 was sequenced using the 50 Mb SureSelect human exome kit (Agilent Technologies), targeting 96.47% of all genes from the NCBI RNA reference collection. The variants were filtered out according to the recessive mode of inheritance (for details, see materials and methods section).
The filtering steps resulted in four potential homozygous variants affecting the genes; HRNR, C4ORF37, WDR41, TMCC3, and four potential compound heterozygous variants in the genes; PLXNA4, PKHD1L1, DPAGT1, and CKAP2 (Table 2). None of the private variants was identified within two homozygous regions determined by homozygosity mapping. Sanger sequencing was used to confirm the exome results and mode of segregation of these variants within the family. As shown by conventional Sanger sequencing, none but one of the variants segregated with the disease phenotype according to an autosomal recessive model of inheritance. The exception was a compound heterozygous change in DPAGT1, which is located at chromosome 11q23.3 (Figure 2). Both affected individuals were compound heterozygous for two missense changes of DPAGT1, c.503T>C (p.L168P) on the maternal allele and c.85A>T (p.I29F), which seemed to be derived from the deceased father (Figure 2). These mutations were highly conserved as indicated by a high phyloP score (conservation score) of 4.9 and 4.8 (Table 2) and the conservation of the amino acids at positions p.Ile29 and p.Leu168 down to the Fruitfly (Figure 3b). We used three bioinformatics based prediction programs such as Polymorphism Phenotyping2 (polyPhen2),16 Sorting Intolerant from Tolerant (SIFT),17 and SNPs&GO,18 to predict the possible causative nature of the identified mutations. The variant c.503T>C was predicted to be likely damaging/deleterious by all three prediction programs, and the variant c.85A>T was predicted to be likely damaging by SIFT and SNPs&GO, while benign by polyPhen2. DPAGT1 encodes a transmembrane protein with 10 transmembrane domains.4 The substituted amino acids are located in the first and fifth transmembrane domains, respectively (Figure 3a), of which the p.L168P mutation introduces a potent helix breaker that will disrupt the helical confirmation of the respective transmembrane domain.19 We have screened 240 ethnically-matched chromosomes for the change c.85A>T and 274 chromosomes for the change c.503T>C. None of the under study mutations was found in the control chromosomes. Both mutations were not present in dbSNP (v135), 1000 genomes data set, and in the Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/).

Structure and location of the DPAGT1 gene on chromosome 11q23.3. The position of the two missense mutations, c.85A>T and c.503T>C is indicated in exons 1 and 4, respectively. Chromatograms revealing the complete segregation of both variations (shaded) with the phenotype in PKMR13.

Schematic representation of DPAGT1 protein and location of the mutations. (a) The white circles indicate hydrophilic residues, whereas gray circles indicate the hydrophobic residues of the protein. The mutations found in this study are indicated by red circles (p.I29F and p.L168P), located in the first and fifth transmembrane domains of the protein, respectively. The mutations that are already described in literature are shown by purple circles. (b) Positions of the amino-acid substitutions I29F and L168P (indicated by pink bar), which are highly conserved down to the Fruitfly, predicting the disruptive effect on the normal function of the protein.
DPAGT1 is involved in protein N-glycosylation in the endoplasmic reticulum and initiates dolichol-PP-glycan synthesis by formation of GlcNAc-PP-dolichol from dolichol-P. Defects in dolichol-PP-glycan synthesis lead to the CDG type I, characterized by the lack of complete glycan chains on otherwise N-glycosylated proteins. Isoelectric focusing (IEF) of serum transferrin, patient III:3 showed a very mild CDG type I profile with a slight increase of asialo- and disialotransferrin as compared to the control (Figure 4a). To confirm the loss of complete N-glycan chains, high-resolution analysis of immunopurified transferrin was carried out on a LC-C8-chip-QTOF mass spectrometer. After deconvolution of the m/z spectra, peaks were observed with masses 75144.17 and 77351.62 Da, indicating non-glycosylated and mono-glycosylated transferrin, respectively. Semiquantitative analysis resulted in ratios of 0.0143 and 0.0991 for the a-/di-oligosaccharide ratio and the mono-/di-oligosaccharide ratio respectively. Reference ranges were determined from the analysis of 56 controls, which resulted in ranges of 0.000137–0.0104 for the a-/di-oligosaccharide ratio and 0.00535–0.0270 for the mono-/di-oligosaccharide ratio (manuscript in preparation). This clearly shows the loss of complete N-glycan chains (Figure 4b), confirming the CDG-I diagnosis. In view of the lack of fibroblasts for subsequent enzymatic confirmation, we searched the exome sequencing data using a CDG-I gene script as we carried out before in CDG-I genes identification.20 No other genomic variants were identified, further confirming the link between DPAGT1 mutations and loss of complete N-glycans in these two patients.

Glycosylation analysis. (a) Transferrin isofocusing for CDG screening. Zero, 2, 4 indicate, respectively, the asialo-, disialo- and tetrasialo-transferrin isoforms. In patient III:3, an increase of asialo- and disialo-transferrin is visible as compared with the control. (b) C8-chip-QTOF mass spectrum of serum immunopurified transferrin. The gray bar indicates the transferrin protein.
Discussion
We have ascertained an autosomal recessive family with two affected individuals presenting with moderate ID, epilepsy, hypotonia, speech problems, abnormal gait, aggressive behavior (Table 1) and mild dysmorphism because of compound heterozygous mutations in the DPAGT1 gene. The causative nature of these mutations was supported by following: (1) The absence of these variants in a large number of control alleles analyzed in our own center or available in public genetic databases. (2) Both mutations p.I29F and p.L168P are predicted to be likely pathogenic and affect highly conserved amino acids in a transmembrane domains of DPAGT1. (3) Analysis of transferrin N-glycosylation finally confirmed the loss of complete glycan chains, the evidence for a CDG-I type glycosylation defect.
DPAGT1 encodes UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosamine-phosphotransferase, the enzyme which is involved in the initial steps of dolichol-linked oligosaccharide biosynthesis. Homozygous deletion of DPAGT1 in mice was shown to be embryonic lethal, most probably because of the widespread cell death.21 The mechanism(s) by which hypoglycosylation causes ID and dysmorphism is not known. Most recently, the DPAGT1 gene has been described as a target of Wnt/β-catenin signaling pathway, which have multiple vital and crucial roles in development.22 This pathway might explain the brain structural anomalies, which can arise during the morphogenetic process of the brain development.
DPAGT1-CDG is a relatively rare CDG-I subtype with only three clinically described cases.4, 7, 8 Previously described patients showed a severe phenotype consisting of ID, seizures, severe hypotonia and microcephaly. The brain structural abnormalities are described in three patients showing delayed myelination and global atrophy.4, 7, 8 However, in comparison to the reported cases, our patients presented with relatively mild clinical features. The affected individuals in this study are the first examples of an adult presentation with a DPAGT1 defect. For the most frequent CDG-I subtype, PMM2-CDG (formerly CDG-Ia), several adolescent patients have been reported presenting with cerebellar hypoplasia.23, 24 In addition, DPM3-CDG has been reported in a 27-year-old female with muscle dystrophy and dilated cardiomyopathy,25 while DOLK-CDG has recently been associated with non-syndromic dilated cardiomyopathy in patients between 5and 18 years of age.26 Moreover, TUSC3-CDG and MAGT1-CDG are implicated in non-syndromic ID patients, with the age range of 25–62 years.27 It can be anticipated that other CDG-I subtypes will be associated with mild/moderate phenotypes as well and that mutations in CDG-I genes might be more common than thought until now.
Given the rapidly increasing power of sequencing capacity, it is expected that large-scale sequencing protocols, such as exome sequencing and whole-genome sequencing, will be implemented in genetic diagnostic testing within the next years. These high-throughput sequencing analyses typically reveal many DNA variants for which the clinical relevance is not evident, including variants in genes that are known to be involved in CDG subtypes. The expanding clinical spectrum for DPAGT1-CDG and other CDGs will necessitate functional analyses to establish the causative nature of such variants, especially in mild cases with only slightly abnormal screening results. Here, we have used highly sensitive and high-resolution mass spectrometry of serum transferrin for fast and specific confirmation of abnormal protein N-glycosylation. Our study emphasizes to consider a congenital disorder of glycosylation in adult patients with ID.
References
Jaeken J : Congenital disorders of glycosylation. Ann N Y Acad Sci 2010; 1214: 190–198.
Theodore M, Morava E : Congenital disorders of glycosylation: sweet news. Curr Opin Pediatr 2011; 23: 581–587.
Lefeber DJ, Morava E, Jaeken J : How to find and diagnose a CDG due to defective N-glycosylation. J Inherit Metab Dis 2011; 34: 849–852.
Wu X, Rush JS, Karaoglu D et al: Deficiency of UDP-GlcNAc:dolichol phosphate n-acetylglucosamine-1 phosphate transferase (DPAGT1) causes a novel congenital disorder of glycosylation type Ij. Hum Mutat 2003; 22: 144–150.
Freeze HH : Human disorders in N-glycosylation and animal models. Biochim Biophys Acta 2002; 1573: 388–393.
Vuillaumier-Barrot S : Molecular diagnosis of congenital disorders of glycosylation. Ann Biol Clin (Paris) 2005; 63: 135–143.
Imtiaz F, Al-Mostafa A, Al-Hassnan ZN : Further delineation of the phenotype of congenital disorder of glycosylation DPAGT1-CDG (CDG-Ij) identified by homozygosity mapping. JIMD Reports 2011; 2: 107–111.
Wurde AE, Reunert J, Rust S et al: Congenital disorder of glycosylation type Ij (CDG-Ij, DPAGT1-CDG): extending the clinical and molecular spectrum of a rare disease. Mol Genet Metab 2012; 105: 634–641.
Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A : A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res 1989; 17: 8390.
Nannya Y, Sanada M, Nakazaki K et al: A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res 2005; 65: 6071–6079.
Hoischen A, Gilissen C, Arts P et al: Massively parallel sequencing of ataxia genes after array-based enrichment. Hum Mutat 2010; 31: 494–499.
Gilissen C, Arts HH, Hoischen A et al: Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet 2010; 87: 418–423.
Rozen S, Skaletsky H : Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–386.
de JG, van Noort WL, van Eijk HG : Optimized separation and quantitation of serum and cerebrospinal fluid transferrin subfractions defined by differences in iron saturation or glycan composition. Adv Exp Med Biol 1994; 356: 51–59.
Babovic-Vuksanovic D, O'Brien JF : Laboratory diagnosis of congenital disorders of glycosylation type I by analysis of transferrin glycoforms. Mol Diagn Ther 2007; 11: 303–311.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Ng PC, Henikoff S : SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31: 3812–3814.
Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R : Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat 2009; 30: 1237–1244.
Alias M, yuso-Tejedor S, Fernandez-Recio J, Cativiela C, Sancho J : Helix propensities of conformationally restricted amino acids. Non-natural substitutes for helix breaking proline and helix forming alanine. Org Biomol Chem 2010; 8: 788–792.
Timal S, Hoischen A, Lehle L et al: Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum Mol Genet 2012; 21: 4151–4161.
Marek KW, Vijay IK, Marth JD : A recessive deletion in the GlcNAc-1-phosphotransferase gene results in peri-implantation embryonic lethality. Glycobiology 1999; 9: 1263–1271.
Sengupta PK, Bouchie MP, Kukuruzinska MA : N-glycosylation gene DPAGT1 is a target of the Wnt/beta-catenin signaling pathway. J Biol Chem 2010; 285: 31164–31173.
Coman D, McGill J, MacDonald R et al: Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci 2007; 14: 668–672.
Vermeer S, Kremer HP, Leijten QH et al: Cerebellar ataxia and congenital disorder of glycosylation Ia (CDG-Ia) with normal routine CDG screening. J Neurol 2007; 254: 1356–1358.
Lefeber DJ, Schonberger J, Morava E et al: Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet 2009; 85: 76–86.
Lefeber DJ, Brouwer AP, Morava E et al: Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet 2011; 7: e1002427.
Molinari F, Foulquier F, Tarpey PS et al: Oligosaccharyltransferase-subunit mutations in nonsyndromic mental retardation. Am J Hum Genet 2008; 82: 1150–1157.
Acknowledgements
We are grateful to all the family members of PKMR13 for the participation in this study. The research work leading to these results has received funding from the European Union's Seventh Framework Program under grant agreement number 241995, project GENCODYS. Z.I was supported by the Higher Education Commission (HEC), Islamabad, Pakistan. The mass spectrometry work was funded by an NWO Medium Investment with a grant number 40-00506-98-9001 to D.J.L.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Iqbal, Z., Shahzad, M., Vissers, L. et al. A compound heterozygous mutation in DPAGT1 results in a congenital disorder of glycosylation with a relatively mild phenotype. Eur J Hum Genet 21, 844–849 (2013). https://doi.org/10.1038/ejhg.2012.257
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.257
Keywords
This article is cited by
-
Glycosylation and behavioral symptoms in neurological disorders
Translational Psychiatry (2023)
-
Clinical glycomics for the diagnosis of congenital disorders of glycosylation
Journal of Inherited Metabolic Disease (2018)
-
Clinical utility gene card for: DPAGT1 defective congenital disorder of glycosylation
European Journal of Human Genetics (2015)
-
Exoom-sequencing in de diagnostiek van ontwikkelingsachterstand/verstandelijke beperking
Tijdschrift voor Kindergeneeskunde (2014)
