
Abstract
Meckel–Gruber syndrome (MKS, OMIM #249000) is a multiple congenital malformation syndrome that represents the severe end of the ciliopathy phenotypic spectrum. Despite the relatively common occurrence of this syndrome among Arabs, little is known about its genetic architecture in this population. This is a series of 18 Arab families with MKS, who were evaluated clinically and studied using autozygome-guided mutation analysis and exome sequencing. We show that autozygome-guided candidate gene analysis identified the underlying mutation in the majority (n=12, 71%). Exome sequencing revealed a likely pathogenic mutation in three novel candidate MKS disease genes. These include C5orf42, Ellis–van-Creveld disease gene EVC2 and SEC8 (also known as EXOC4), which encodes an exocyst protein with an established role in ciliogenesis. This is the largest and most comprehensive genomic study on MKS in Arabs and the results, in addition to revealing genetic and allelic heterogeneity, suggest that previously reported disease genes and the novel candidates uncovered by this study account for the overwhelming majority of MKS patients in our population.
Similar content being viewed by others


Introduction
Meckel–Gruber syndrome (MKS, OMIM #249000) is a multiple congenital malformation syndrome characterized mainly by early lethality, occipital encephalocele, dysplastic kidneys, and polydactyly, but the phenotype also includes liver fibrosis, microphthalmia, cleft palate, and a wide-array of associated brain anomalies.1 MKS tends to be a rare disease in most populations (<1:20 000) with the notable exception of Finland where a birth prevalence of 1:9000 was estimated, and Arabia where Teebi and Teebi reported a much higher frequency of 1:3500.2, 3
Our understanding of MKS pathogenesis has improved greatly over the past few years since it was first revealed that MKS1, the first gene found to be mutated in MKS, encodes a ciliary protein.4 Subsequently, nine additional genes have been identified, all similarly encoding ciliary proteins (TMEM216 (MKS2), TMEM67 (MKS3), CEP290 (MKS4), RPGRIP1L (MKS5), CC2D2A (MKS6), NPHP3 (MKS7), TCTN2 (MKS8), B9D1 (MKS9) and B9D2 (MKS10)).4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 The finding that defective ciliary biology is the core molecular pathology of MKS made it possible to dissect the pathogenesis of each of its manifestations. For instance, the primary cilium plays a critical role in SHH signaling that controls anterior–posterior and dorsal–ventral patterning of the developing limb buds and neural tube, respectively, thus explaining the polydactyly and neural tube defects that characterize MKS at the molecular level. Similarly, the third classical feature of cystic dysplasia of the kidneys is likely caused by abnormal patterning secondary to defective urine flow-induced signaling that is normally transduced by the cilia on the luminal surface of renal epithelium.
Defective ciliary biology is not unique to MKS; an expansive group of disorders commonly known as ciliopathies share this fundamental pathological feature.15 In fact, mutations in MKS genes have been reported to cause other ciliopathies, for example, MKS1 and Bardet–Biedl syndrome,16 TMEM216 and Joubert syndrome,17 TMEM67 and Joubert syndrome and nephrophthisis,18, 19 CEP290 in non-syndromic retinal dystrophy, Senior–Loken syndrome, nephronophthisis, Joubert syndrome, and Bardet–Biedl syndrome,20 RPGRIP1L and Joubert syndrome,21 and CC2D2A and Joubert syndrome.22 The factors that determine the ultimate clinical phenotype are not completely understood but there is growing evidence that ciliopathies represent a spectrum of clinical severity that correlates to some extent with the severity of the ciliary defect. Consistent with MKS being at the severe end of the clinical spectrum, most causative mutations are truncating in nature while hypomorphic mutations in the same genes cause less severe phenotypes.23
The genetic heterogeneity of MKS has yet to be fully captured. Indeed, a recent study showed that only half of the cases can be traced to a mutation in any of the known genes (seven were included in the analysis, so the additional contribution of TCTN2 could not be inferred).14 The autosomal recessive nature of MKS, the highly consanguineous nature of the Arab population where MKS is particularly prevalent, and the success of exome sequencing in other recessive disorders encouraged us to undertake a comprehensive genomic study to fully delineate the genetic architecture of MKS in our population by combining the power of autozygome and exome analysis. In this largest genomic MKS case series in Arabs to date, we show that autozygome-guided candidate gene analysis successfully identified the underlying mutation in >70% of the cases. In the remaining cases, we used exome sequencing followed by autozygome filtration to identify three novel candidate genes, one of which was very recently reported to be also mutated in Joubert syndrome. Assuming the candidate MKS genes that we report here are replicated by future studies, it appears that most of the genetic heterogeneity of MKS, at least in Arabs, has been captured.
Materials and methods
Human subjects
For case definition, we used a relaxed definition of occipital encephalocele as a must-have feature plus any combination of cleft palate, dysplastic kidneys, liver fibrosis, polydactyly, and early lethality. All cases were fully evaluated by an experienced dysmorphologist and/or neonatologist. Eligible cases were recruited after obtaining IRB-approved informed written consent from the parents (KFHSRC RAC#2080006). Blood in EDTA and, when possible, PAXGene tubes was collected for DNA and RNA extraction, respectively.
Autozygome analysis
DNA samples from both affected and unaffected family members were genotyped on Axiom Chip platform as per the manufacture’s protocol (Affymetrix, Santa Clara, CA, USA). Runs of homozygosity (ROH) >2 Mb and that span 107 SNPs were used as surrogates of autozygosity given the consanguineous nature of the families using autoSNPa (http://dna.leeds.ac.uk/autosnpa/). The entire set of autozygosity blocks (autozygome) was determined for each patient. The shared haplotype between parents as well as relatives with similarly affected children was determined by using IBDelphi (http://dna.leeds.ac.uk/ibdelphi/).
Exome sequencing and analysis
Exome capture was performed using TruSeq Exome Enrichment kit (Illumina, San Diego, CA, USA) following the manufacturer’s protocol. Samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using Illumina HiSeq 2000 Sequencer. The reads were mapped against UCSC hg19 (http://genome.ucsc.edu/) by BWA (http://bio-bwa.sourceforge.net/). The SNPs and Indels were detected by SAMTOOLS (http://samtools.sourceforge.net/). The resulting variants were filtered by only considering homozygous novel changes (absent in dbSNP, 1000 Genomes and 200 in-house Saudi exomes) within the autozygome followed by in silico analysis for potential pathogenicity. Variants that survived these filters were then Sanger sequenced in at least 192 normal Saudi controls, that is, 384 chromosomes.
Workflow
As summarized in Figure 1, cases were first subjected to autozygome-guided mutation analysis of known MKS genes. If negative, cases were exome sequenced. Exomes were first examined for potential compound heterozygous mutations in known MKS genes since these will be missed by the autozygome approach. If negative, we proceeded with the filtration scheme outlined above (see Exome Sequencing).

Workflow of the current study.
Results
Human subjects
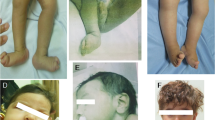
In total, 18 families that met the inclusion criteria were recruited, all consanguineous. All pedigrees are shown in Supplementary Figure S1 and representative clinical pictures for some of the MKS patients are shown in Figure 2. Table 1 summarizes their clinical features. One family was initially recruited despite lack of additional features of MKS besides occipital encephalocele because there were no ultrasonographic features suggestive of another diagnosis and the fetus was unavailable for clinical assessment after delivery. This case was later found to have two base pair deletion in POMT1 gene (NM_007171.3:c.2179_2180delTC), so it was excluded and is not considered further in this study although it serves as a reminder that encephalocele is a birth defect with a broad differential diagnosis.24 As expected, our relaxed clinical definition resulted in the recruitment of two cases (MKS_F1 and MKS_F15) that do not meet the classical minimal definition of early lethality, encephalocele, and cystic/dysplastic kidneys. However, we opted to retain these cases in our molecular analysis to explore the extent of variable expressivity in MKS especially when these cases were subsequently found to harbor mutations in genes recently reported to cause Joubert syndrome (see below).

(a–c) Facial features of some MKS patients MKS_F1, F2, and F8, respectively, showing: microcephaly, sloping forehead, hypertelorism, micrognathia, potter-like facies, and severe occipital encephalocele (black arrows). (d, e) Representative hand and foot images showing postaxial polydactyly in MKS_F2. (f, g) Antenatal ultrasound for MKS_F12 patient showed encephalocele (white arrow) and enlarged polycystic kidney (dashes arrows).
Autozygome-guided mutational analysis
Consistent with all families being consanguineous, all index cases had evidence of autozygosity and the percentage of their genomes represented by the autozygome ranged 2.5–18.4% (Supplementary Table S1). At the time of the study, 10 MKS disease genes were identified (TMEM67, TMEM216, CEP290, CC2D2A, NPHP3, RPGRIP1L, TCTN2, B9D1, and B9D2). Gene(s) that overlap with autozygome were Sanger sequenced and this led to the identification of a likely pathogenic homozygous mutation in 9 of 17 families (Figure 1). Supplementary Figure S2 shows both the multi-species alignment of orthologs as well as the PolyPhen/SIFT scores for each of the novel missense variants. For three families (MKS_F4, MKS_F11, and MKS_F13), we had no access to the index so we used IBDelphi to infer the shared ancestral haplotype between the parents which helped us identify the following pathogenic mutations: c.1506-2A>G in TCTN2 (NM_024809.3), c.1855_1858del in CEP290 (NM_025114) and c.613C>T; p.R205* in CEP290 (NM_025114.3), respectively. Thus autozygome-guided mutational analysis and analysis to the shared haplotype between parents led to the successful identification of the underlying mutation in 71% of cases (Table 2; Figure 3).

Sequence chromatograms of novel mutations listed in Table 2. Mutation sites are denoted with asterisks or lines.
Autozygome-filtered exome sequencing
Five families in which autozygome-guided mutation analysis failed to identify the causative mutation in our initial list of 10 genes were exome sequenced (Figure 1). Summary of the depth of coverage and the amount of exome covered are shown in Supplementary Table S2. Variants were excluded when present homozygous in Saudi controls and heterozygous at a frequency >1% ie present twice in 200 exomes. The filtration steps used in interpreting the exome results are summarized in Supplementary Table S3 and the novel variants that survived the various filters are listed in Table 3. MKS_F1 was found to harbor a novel homozygous mutation that abolished a consensus donor site in TMEM237 (c.869+1G>A) and RT–PCR confirmed the resulting exonic skipping, which predicts in-frame deletion of >60 amino acids p(Ap.Met227_Arg291del). Mutations in this gene have recently been reported to cause a phenotypic spectrum between Joubert syndrome and MKS.25 MKS_F15 was found to harbor the same homozygous truncating mutation we recently reported in a gene that causes Joubert syndrome (C5orf42).26, 27 Two exomes (MKS_16 and MKS_2) highlighted novel MKS candidate genes: EXOC4 and EVC2, respectively, whereas exome sequencing of MKS_F18 failed to generate any compelling candidate variants after applying the various filters (Figure 1; Supplementary Table S4).
Discussion
Although many MKS genes were mapped using consanguineous Arab families, the mutation distribution of these genes in Arabs is unknown but deserves investigation to inform clinical sequencing and genetic counseling. Furthermore, the success we have had in combining autozygome and exome analysis in delineating the genetic architecture of other genetically heterogeneous disorders, for example, Osteogenesis imperfecta, retinal dystrophy, cataract, mitochondrial diseases, Bardet–Biedl syndrome, and Joubert syndrome26, 28, 29, 30, 31 (and Abu Safieh et al (2012)32, Shaheen et al (2012)33), encouraged us to use a similar approach on MKS to not only determine the mutation distribution in known MKS disease genes but to also potentially identify novel candidate disease genes.
To address the first aim, that is, determination of the mutation distribution in known MKS genes, we took advantage of the consanguineous nature of our study population to trace causative mutations by virtue of the autozygosity signature that typically characterizes identical-by-descent recessive mutations. Indeed, we show that autozygome-guided mutational analysis has efficiently identified the causative variant in each family in which MKS is caused by a mutation in a known gene. Importantly, even though the mutations in TMEM237 and C5orf42 were identified by exome sequencing (these were unknown to cause MKS at the time of the analysis), both could have easily been identified using the same approach further increasing the yield of this approach to 88% (15 out of 17) in the setting of MKS. This is consistent with the trend we and others have shown in terms of the power of this approach in the setting of genetically heterogeneous autosomal recessive diseases.28, 29, 34
In order to explore the potential of revealing novel disease genes, all cases in which autozygome-guided mutational analysis was negative were exome sequenced. As we have shown previously, autozygome served as an extremely powerful filter of the resulting variants.35, 36 In Family MKS_F1, two variants survived filtration but TMEM237 was recently described as a novel Joubert gene.25 Some of the original reported cases had occipital encephalocele and cystic kidney so it seems likely that our mutation is the causal variant.
In Family MKS_F15, only two variants survived the various filters (C5orf42, NM_023073.3; c.7988_7989delGA and FAM48A, NM_001014286; c.236A>G). We could not determine which of the two is likely to be causal because while the former is truncating in nature, the latter missense mutation affected a gene that is clearly linked to neural tube defects in mice.37 During the preparation of this manuscript, we and others identified several truncating mutations in C5orf42 in the context of Joubert syndrome.26, 27 Since MKS is a more severe phenotype, it is tempting to speculate that the additional allele in FAM48A may have influenced the severity of the phenotype. However, we caution against the overinterpretation of the presence of the FAM48A allele because occipital encephalocele/meningocele has been previously reported in the setting of C5orf42 mutations.26, 27 Indeed, we note that case MKS_F15, while meeting our operational clinical definition of MKS, did not have polydactyly or renal involvement so it can be argued that even if C5orf42 is indeed an MKS disease gene, it causes atypical forms of the disease. It is worth highlighting that the only two cases in our series that did not meet the classical definition of MKS are those with mutations in TMEM237 and C5orf42, which have been shown to cause both classical Joubert syndrome as well as cases that are Meckel-like as we explained above. Only through analysis of the full extent of C5orf42 and TMEM237 contribution to MKS using large case series will it be apparent if classical MKS can also be caused by mutations in these genes.
Families MKS_F2 and MKS_F16 deserve special emphasis. In Family MKS_F2, the child had all the classical features of MKS and yet his exome sequencing revealed a novel variant in EVC2, a known disease gene for Ellis–van-Creveld syndrome. Although the mutation is in-frame, it inserts eight amino acids p.Lys1293_Lys1300dup. Interestingly, mutations in EVC2 have been shown to modulate SHH signaling. Furthermore, EVC2 has been found recently to localize at the basal body of the primary cilium so it is tempting to speculate that this may be the link between EVC2 and MKS.38, 39, 40 Thus, our results are build on recent data to suggest that EVC is a ciliopathy. However, we note that lack of EVC findings on skeletal survey and fetal echocardiography in this case strongly argue that this is a bona fide MKS phenotype rather than a simple clinical overlap between EVC and MKS as was once reported.41 In Family MKS_F16, the variant we identified in EXOC4 (also known as SEC8) is particularly interesting. The gene encodes one of the several exocyst proteins that are recruited to the basal body of budding cilia along with the members of disheveled family of proteins in order to dock the basal body to the membrane and allow the nascent cilia to form.42, 43 Knockdown of another exocyst gene SEC10 was found to impair ciliogenesis, raising the possibility that defective SEC8 may lead a similar cellular phenotype. Furthermore, another exocyst gene EXOC8 was recently found mutated in a patient with Joubert syndrome.44 Of note, the variant we identified is conserved down to Fugu and zebrafish.
One of the cases (MKS_F18) was not solved by Exome sequencing. In our experience, only around 70% of autosomal recessive cases are solved by this techniques and this could be explained by various reasons. First, the mutation may not be in the exon or exon/intron boundary. Second, the mutation may be in an exon that has not been annotated and thus not included in the capture design. Third, the mutation may belong to a class that is particularly difficult to assay using next-generation sequencing (eg, genomic rearrangements, repeats, etc). Fourth, the mutation may simply have been skipped by either the capture or sequencing reaction.
Of the three proposed novel MKS candidate genes, C5orf49 has been independently confirmed through recent reports of Joubert syndrome patients with biallelic mutations in these genes.26, 27 EVC2 and EXOC4, however, will have to await future confirmation as bona fide MKS loci. If independently confirmed, our study shows that, at least in Arabs, most of the genetic heterogeneity of this disorder has been captured and that the contribution of future MKS loci is likely to be minimal. One previous study showed that seven MKS genes (TMEM67, TMEM216, CEP290, CC2D2A, B9D1, and RPGRIP1L) were found to be biallelically mutated in 54% of their cohort, a higher contribution for these particular genes than what we observed in our study sample (32%). Thus, it seems possible that the higher percentage of mutation-positive MKS cases (94% by including TCTN2, TMEM237, C5orf42, and if EVC2 and EXOC4 are confirmed by future reports) may potentially be relevant to other populations as well.
In summary, we show in the largest genomic study on an MKS cohort the mutation distribution among Arabs where the disease is particularly prevalent. This genomic approach revealed interesting novel candidates that may potentially bring, along with recently described MKS disease genes, the quest to unravel the genetics of MKS to its final stages making it possible for the overwhelming majority of these patients to receive genetic counseling that is informed by molecular confirmation.
References
Seller MJ : Phenotypic variation in Meckel syndrome. Clin Genet 1981; 20: 74–77.
Teebi AS, Teebi SA : Genetic diversity among the Arabs. Community Genet 2005; 8: 21–26.
Salonen R, Norio R : The Meckel syndrome in Finland: epidemiologic and genetic aspects. Am J Med Genet 1984; 18: 691–698.
Kyttala M, Tallila J, Salonen R et al: MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet 2006; 38: 155–157.
Consugar MB, Kubly VJ, Lager DJ et al: Molecular diagnostics of Meckel-Gruber syndrome highlights phenotypic differences between MKS1 and MKS3. Hum Genet 2007; 121: 591–599.
Dawe HR, Smith UM, Cullinane AR et al: The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet 2007; 16: 173–186.
Valente EM, Logan CV, Mougou-Zerelli S et al: Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet 2010; 42: 619–625.
Smith UM, Consugar M, Tee LJ et al: The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet 2006; 38: 191–196.
Baala L, Audollent S, Martinovic J et al: Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet 2007; 81: 170–179.
Frank V, den Hollander AI, Bruchle NO et al: Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum Mutat 2008; 29: 45–52.
Delous M, Baala L, Salomon R et al: The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet 2007; 39: 875–881.
Tallila J, Jakkula E, Peltonen L, Salonen R, Kestila M : Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet 2008; 82: 1361–1367.
Shaheen R, Faqeih E, Seidahmed MZ et al: A TCTN2 mutation defines a novel Meckel Gruber syndrome locus. Hum Mutat 2011; 32: 573–578.
Hopp K, Heyer CM, Hommerding CJ et al: B9D1 is revealed as a novel Meckel syndrome (MKS) gene by targeted exon-enriched next-generation sequencing and deletion analysis. Hum Mol Genet 2011; 20: 2524–2534.
Nigg EA, Raff JW : Centrioles, centrosomes, and cilia in health and disease. Cell 2009; 139: 663–678.
Leitch CC, Zaghloul NA, Davis EE et al: Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet 2008; 40: 443–448.
Edvardson S, Shaag A, Zenvirt S et al: Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am J Hum Genet 2010; 86: 93–97.
Baala L, Romano S, Khaddour R et al: The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet 2007; 80: 186–194.
Otto EA, Tory K, Attanasio M et al: Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet 2009; 46: 663–670.
Coppieters F, Lefever S, Leroy BP, De Baere E : CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat 2010; 31: 1097–1108.
Arts HH, Doherty D, van Beersum SE et al: Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet 2007; 39: 882–888.
Gorden NT, Arts HH, Parisi MA et al: CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet 2008; 83: 559–571.
Salonen R, Kestila M, Bergmann C : Clinical utility gene card for: Meckel syndrome. Eur J Hum Genet 2011; 19: ; e-pub ahead of print 2 February 2011; doi:10.1038/ejhg.2010.255.
Cohen MM, Lemire RJ : Syndromes with cephaloceles. Teratology 1982; 25: 161–172.
Huang L, Szymanska K, Jensen VL et al: TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet 2011; 89: 713–730.
Alazami AM, Alshammari MJ, Salih MA et al: Molecular characterization of Joubert syndrome in Saudi Arabia. Hum Mutat 2012; 33: 1423–1428.
Srour M, Schwartzentruber J, Hamdan FF et al: Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am J Hum Genet 2012; 90: 693–700.
Shamseldin HE, Alshammari M, Al-Sheddi T et al: Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J Med Genet 2012; 49: 234–241.
Abu Safieh L, Aldahmesh MA, Shamseldin H et al: Clinical and molecular characterisation of Bardet-Biedl syndrome in consanguineous populations: the power of homozygosity mapping. J Med Genet 2010; 47: 236–241.
Abu-Safieh L, Al-Anazi S, Al-Abdi L et al: In search of triallelism in Bardet-Biedl syndrome. EJHG 2012; 20: 420–427.
Aldahmesh MA, Khan AO, Mohamed JY et al: Genomic analysis of pediatric cataract in Saudi Arabia reveals novel candidate disease genes. Genet Med 2012.
Abu-Safieh L, Alrashed M, Anazi S et al: Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res 2012, ; e-pub ahead of print 26 October 2012.
Shaheen R, Alazami AM, Alshammari MJ et al: Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J Med Genet 2012; 49: 630–635.
Shaheen R, Al-Dirbashi OY, Al-Hassnan ZN et al: Clinical, biochemical and molecular characterization of peroxisomal diseases in Arabs. Clin Genet 2011; 79: 60–70.
Shaheen R, Faqeih E, Sunker A et al: Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am J Hum Genet 2011; 89: 328–333.
Aldahmesh MA, Mohamed JY, Alkuraya HS et al: Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am J Hum Genet 2011; 89: 745–750.
Zohn IE, Li Y, Skolnik EY, Anderson KV, Han J, Niswander L : p38 and a p38-interacting protein are critical for downregulation of E-cadherin during mouse gastrulation. Cell 2006; 125: 957–969.
Blair HJ, Tompson S, Liu YN et al: Evc2 is a positive modulator of Hedgehog signalling that interacts with Evc at the cilia membrane and is also found in the nucleus. BMC Biol 2011; 9: 14.
Valencia M, Lapunzina P, Lim D et al: Widening the mutation spectrum of EVC and EVC2: ectopic expression of Weyer variants in NIH 3T3 fibroblasts disrupts Hedgehog signaling. Hum Mutat 2009; 30: 1667–1675.
Ruiz-Perez VL, Goodship JA : Ellis-van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to hedgehog ligands. Am J Med Genet C Semin Med Genet 2009; 151C: 341–351.
Kemperdick H, Ammermann M, Janssen F, Lange H, Moubayed P : [The differential diagnosis of the Meckel syndrome and the Ellis-van-Creveld syndrome with encephalocele (author’s transl)]. Klin Padiatr 1975; 187: 87–93.
Ganner A, Lienkamp S, Schafer T et al: Regulation of ciliary polarity by the APC/C. Proc Natl Acad Sci USA 2009; 106: 17799–17804.
Park TJ, Mitchell BJ, Abitua PB, Kintner C, Wallingford JB : Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat Genet 2008; 40: 871–879.
Dixon-Salazar TJ, Silhavy JL, Udpa N et al: Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med 2012; 4: 138ra178.
Acknowledgements
We thank the study families for their enthusiastic participation. We thank Dr Mohammed Al-Balawi for his help in extracting and storing DNA samples. We also thank the Genomic and Sequencing Core Facilities at KFSHRC for their technical help. This study was funded in part by KACST Grant 09-MED941-20 (FSA) and DHFMR Collaborative Research Grant (FSA).
Author contributions
Ranad Shaheen and Fowzan S Alkuraya: collected and analyzed the data and wrote the manuscript. Eissa Faqeih, Muneera J Alshammari, Abdulrahman Swaid, Lihadh Al-Gazali, Elham Mardawi, Shinu Ansari, Mohammed Z Seidahmed, Muhammed Mutairi, Chantal Farra, Wesam Kurdi, and Shatha Al-Rasheed: collected and analyzed the data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Shaheen, R., Faqeih, E., Alshammari, M. et al. Genomic analysis of Meckel–Gruber syndrome in Arabs reveals marked genetic heterogeneity and novel candidate genes. Eur J Hum Genet 21, 762–768 (2013). https://doi.org/10.1038/ejhg.2012.254
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.254
Keywords
This article is cited by
-
The exocyst complex in neurological disorders
Human Genetics (2023)
-
A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies
Nature Genetics (2018)
-
Clinical utility gene card for: Meckel syndrome – update 2016
European Journal of Human Genetics (2016)
-
The many faces of KIF7
Human Genome Variation (2015)
-
C5orf42 is the major gene responsible for OFD syndrome type VI
Human Genetics (2014)
