
Abstract
XPD (ERCC2) is a DNA helicase involved in nucleotide excision repair and in transcription as a structural bridge tying the transcription factor IIH (TFIIH) core with the cdk-activating kinase complex, which phosphorylates nuclear receptors. Mutations in XPD are associated with several different phenotypes, including trichothiodystrophy (TTD), with sulfur-deficient brittle hair, bone defects, and developmental abnormalities without skin cancer, xeroderma pigmentosum (XP), with pigmentary abnormalities and increased skin cancer, or XP/TTD with combined features, including skin cancer. We describe the varied clinical features and mutations in nine patients examined at the National Institutes of Health who were compound heterozygotes for XPD mutations but had different clinical phenotypes: four TTD, three XP, and two combined XP/TTD. We studied TFIIH-dependent transactivation by nuclear receptor for vitamin D (VDR) and thyroid in cells from these patients. The vitamin D stimulation ratio of CYP24 and osteopontin was associated with specific pairs of mutations (reduced in 5, elevated in 1) but not correlated with distinct clinical phenotypes. Thyroid receptor stimulation ratio for KLF9 was not significantly different from normal. XPD mutations frequently were associated with abnormal VDR stimulation in compound heterozygote patients with TTD, XP, or XP/TTD.
Similar content being viewed by others

Introduction
Trichothiodystrophy (TTD) (MIM no. 601675) is a rare autosomal recessive multisystem disorder characterized by sulfur-deficient brittle hair, neurological abnormalities, ichthyosis, and cutaneous photosensitivity without skin cancer.1 TTD may be manifested when both alleles are affected by mutations in one of the three DNA repair genes (XPB (ERCC3) MIM no. 133510, XPD (ERCC2) MIM no. 126340, TTDA (GTF2H5) MIM no. 608780),2 or in TTDN1 (C7orf11) MIM no. 609188,3 a gene of unknown function. Interestingly, mutations in XPB and XPD can also cause xeroderma pigmentosum (XP) (MIM no. 278730),4, 5, 6, 7 with sunlight-induced freckle-like skin pigmentation and >10 000-fold increased risk of skin cancer.8 TTD and XP have an incidence of about 1 per million live births in the United States and Western Europe.9 Very rare XP/TTD patients have clinical features of both XP and TTD10, 11 and carry an increased cancer risk.
The XPD, XPB, and TTDA proteins are associated with transcription factor IIH (TFIIH), a 10 protein complex involved in both initiation of transcription by RNA polymerase II and nucleotide excision repair.12, 13, 14, 15 XP and TTD patients may have different, although overlapping, XPD mutations, suggesting that the site of mutation influences the clinical phenotype.10, 16, 17, 18, 19, 20 Cells from XP and TTD patients with XPD mutations differ in the rate of repair of ultraviolet-induced DNA photoproducts and in the time course of localization and removal of nucleotide excision repair proteins to sites of UV damage.10
Nuclear receptors are hormone-dependent transcription factors that play essential roles in development, differentiation, and metabolism by controlling the expression of specific networks of genes.21, 22 XPD mutations reduce basal transcription activity in TTD cells and inhibit nuclear receptor transactivation in XP as well as in TTD cells.23, 24, 25 Using our unique collection of fibroblast strains established from the TTD, XP, and XP/TTD patients we examined at the National Institutes of Health (NIH) over the past three decades,8, 26, 27, 28 we selected cells from nine patients with XPD mutations (four TTD, two XP/TTD, and three XP), with varied and distinct clinical features. We tested two types of nuclear receptor, vitamin D receptor (VDR) and thyroid receptor, which are important for bone growth and maintenance, development, and innate and adaptive immunity.21, 22 We hypothesized that these patients would have abnormal nuclear receptor transactivation.
Materials and methods
Clinical protocol, cells, maintenance culture conditions, and mutation analysis
Patients were studied under protocol 99-C-0099 approved by the National Cancer Institute Institutional Review Board, which adhered to the Declaration of Helsinki Principles. Skin biopsies were obtained and sent to the Human Genetic Mutant Cell Repository (Camden, NJ, USA) or to NCI-Frederick (Frederick, MD, USA) for establishment of fibroblast cell cultures. Fibroblast cultures from four TTD, two XP/TTD, three XP patients (Table 1), and normal controls AG04438 and AG13145 were grown in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) containing 4 mM glutamine and 10% fetal calf serum (FCS) (Hyclone Laboratories Inc., Logan, UT, USA) in an 8% CO2 humidified incubator at 37 °C as described previously.10 Mutation analysis was performed as described previously.10 The XPD GenBank sequences used were NM_0000400.3 (XPD cDNA) with numbering based on +1 as the A of the ATG initiation codon and NP_000391.1 (XPD protein).
Hormone stimulation and DNA/RNA isolation
Nuclear receptor activation was performed as described previously.25 In brief, fibroblasts were plated at 50–60% confluence in complete media for 24 h. The media were changed to a phenol red-free media containing 10% charcoal-treated FCS and the cells were incubated for 12 h with either 1,25-dihydroxyvitamin D3 (1,25(OH)2D3; VD) (Sigma-Aldrich, St Louis, MO, USA) or triiodothyronine (T3) (Sigma-Aldrich). Total cytoplasmic RNA was isolated from cells using the RNAqueous small-scale phenol-free total RNA isolation kit (Ambion Inc., Austin, TX, USA). DNA was isolated using DNAzol reagent (Invitrogen, Carlsbad, CA, USA).
Quantitative real-time RT–PCR
Quantitative real-time RT-PCR (qPCR) was performed as described.25 Total RNA was treated with DNA-free DNase (Ambion Inc., Austin, TX, USA) and then converted to cDNA using Superscript II (Invitrogen, Carlsbad, CA, USA) as described.29 A measure of 2 μl (corresponding to 25 ng RNA) of RT reaction was used as template for each PCR reaction. qPCR assays were carried out on a Bio-Rad iCycler iQ system (Bio-Rad Laboratories Inc., Hercules, CA, USA) using intercalation of SYBR Green (iQ SYBR Green Supermix; Bio-Rad) as the fluorescence reporter. Two-step PCR (denaturation at 95 °C for 15 s and annealing/extension at 58 °C for 1 min) was performed with data collection and analysis during the combined annealing and extension step. Melt-curve analyses were performed on all PCR reactions to rule out nonspecific amplification. The following forward and reverse primer sequences were used for the target genes: CYP24 MIM no. 126065 (VD target gene) – 5′-TCTTGACAAGGCAACAGTTC-3′, 5′-AAGCCAACGTTCAGGTCTAA-3′ (PCR fragment: 132 bp); osteopontin (OPN) (SPP1) MIM no. 166490 (VD target gene) – 5′-TATGATGGCCGAGGTGATAG-3′, 5′-AGGTGATGTCCTCGTCTGTA-3′ (PCR fragment: 86 bp); KLF9 MIM no. 602902 (T3 target gene) – 5′-CCAAGAGCTTGTTGGACCTGAACA-3′, 5′-TCGGATCCCATATCCTCATCTGGACT-3′ (PCR fragment: 96 bp); intercellular adhesion molecule 1 (ICAM1) MIM no. 147840 (not a VD target gene) – 5′-TTTTCTATCGGCACAAAAGC-3′, 5′-AATGCAAACAGGACAAGAGG-3′ (PCR fragment: 232 bp) (product number VHPS-4429 from RealTimePrimers.com); and 18S RNA (internal control) – 5′-CGGACAGGATTGACAGATTG-3′, 5′-TGCCAGAGTCTCGTTCGTTA-3′ (PCR fragment: 98 bp). Ct values were normalized relative to 18S RNA expression.
Data analysis and statistics
The results are the mean of at least two independent experiments performed with duplicate samples. All data are presented as fold change against each control (specified as either mean of non-stimulated cells or mean of normal cells under the same conditions). Student’s t-test with a two-tailed distribution was used to analyze the data. Statistical significance was judged to be a P-value of <0.05.
Results
Clinical features differ markedly among TTD, XP, and XP/TTD patients with XPD mutations
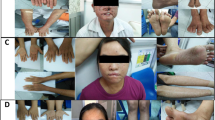
Patient TTD354BE was a Caucasian female with TTD (Figures 1a–c) who was compound heterozygous for a previously unreported XPD mutation (p.G459-T460ins34aa) and p.A725T, which was present in another TTD patient (TTD421BE)30 (Table 1 and Figure 2). TTD354BE had features of severe TTD,1, 31 including tiger tail banding of her hair (Figure 1c), short stature (<3%-ile), congenital cataracts, microcephaly, brain dysmyelination, global developmental delay, including central osteosclerosis and peripheral osteopenia of her bones, elevated hemoglobin A2 (HbA2) levels, and a low mean corpuscular volume. She had recurrent ear infections and pneumonias and died at the age of 9 years from acute infection following hip surgery.27

TTD and XP patients studied. (a) TTD patient TTD354BE at age 3 years. She has extremely short stature (70 cm, <3%-ile), diminutive features, and cheerful affect, typical of TTD. She was unable to stand without assistance. Note relative size of her parent’s hand. (b) TTD patient TTD354BE at age 7 years. Her hair is extremely brittle and breaks easily. She never needs a haircut. There is loss of the lateral portion of her eyebrows. Ichthyosis is present on her back (arrow) and chest. (c) TTD patient TTD354BE hair. Examination with polarized microscopy shows prominent alternating dark and light ‘tiger tail’ banding characteristic of TTD (arrows) and trichoschisis defects of the hair shaft (*). (d) TTD patient TTD412BE at age 7 years. He had short stature (96 cm, <3%-ile) and a cheerful affect. He was unable to stand without assistance. He utilized a g-tube (arrow) for feeding. (e) TTD patient TTD412BE has long hair, micrognathia (small jaw), and mild ichthyosis (arrow) with cheerful affect. (f) XP patient XP29BE at age 21 years. He has freckle-like pigmented lesions on his chest, neck, and face. There are numerous scars on his face from multiple surgical procedures for removal of skin cancers. Several lesions suspicious for skin cancer are present (circles). Note pterygium in the lateral conjunctiva of his right eye. (g) XP patient XP34BE at age 24 years. Despite severe acute sun sensitivity, he has used sun protection and shows minimal pigmentary changes of his skin.

XPD domains, functional helicase motifs, and mutation sites. Relative positions of the XPD domains are shown above a linear schematic of the human XPD protein. The XPD catalytic core of Sulfolobus acidocaldarius XPD (SaXPD) is comprised of four domains: two Rad51/RecA-like helicase domains (HD1 and HD2) with two additional domains (4FeS and Arch domains) inserted into HD1.18 The helicase motifs (labeled with Roman numerals) are conserved from SaXPD to human XPD.18 However, human XPD has an extended C-terminal domain (CTD) that has been proposed to be the TFIIH p44 interface.23 The relative positions of the mutations found in XP patients (XP17BE, XP29BE, and XP34BE), TTD patients (TTD351BE,TTD354BE, TTD404BE, andTTD412BE), and XP/TTD patients (XPTTD268BE and XPTTD306BE) are shown. XP patient mutations are presented above the protein schematic, and TTD patient mutations and XP/TTD patient mutations are presented below.
Patient TTD412BE was a 7-year-old Caucasian male with TTD (Figure 1d and e)27 with p.A725P mutation in one XPD allele and p.L461V plus p.V716-R730del in the other allele, resulting from a g.18339C>G splice mutation (Table 1 and Figure 2). These same mutations have been reported previously in another patient (TTD183ME).32 Interestingly, while TTD183ME was acutely sun sensitive, TTD412BE did not have this same sun sensitivity.
Patient TTD351BE (Table 1 and Figure 2) was a Caucasian male with a unique p.R378H mutation and the TTD-specific mutation, p.R722W.20 A mouse engineered to have this mutation was shown to have some clinical features of TTD.33 The TTD351BE cells had normal DNA repair.10 The patient died at the age of 15 years owing to infection and respiratory failure following hip surgery.27
Patient TTD404BE27 was a 11-year-old Caucasian male with a novel p.R112C mutation at the same site as a more common p.R112H mutation reported in other TTD patients11, 16 (Table 1 and Figure 2). He also had a p.D681N mutation, which was present in a patient with cerebro-ocular-facial-skeletal (COFS) syndrome34 and in XP patient XP17BE.10 He had long hair with tiger tail banding under polarized microscopy.
Patient XPTTD268BE was a 25-year-old Caucasian male who had the commonly reported TTD mutation, p.R112H, and a unique p.C663R mutation10 (Table 1 and Figure 2). He had long hair and TTD features of tiger tail banding on polarized microscopy, ichthyosis, cataracts, leukodystrophy, and recurrent infections. He also had XP-type freckle-like pigmentation supporting the diagnosis of XP/TTD.11
Patient XPTTD306BE was a 14-year-old Caucasian male with XP/TTD who had a novel mutation, p.S51F, and a commonly reported TTD-specific mutation, p.R722W, in the XPD protein10 (Table 1 and Figure 2). He had XP features of acute sun sensitivity and freckle-like skin pigmentation but no skin cancers. In addition, he had the characteristic TTD features of ‘tiger tail’ banding of his hair on polarized microscopy, ichthyosis, short stature, microcephaly, and brain dysmyelination and recurrent pneumonias.6
Patient XP17BE10 was an African-American male with the p.R683W mutation commonly seen in XP patients,10, 17, 19, 20 as well as in XP29BE and XP34BE. He also had a p.D681N mutation, which was present in a COFS patient34 and in a TTD patient TTD404BE (Table 1 and Figure 2).
Patient XP29BE was a Caucasian male with severe XP phenotype with neurological abnormalities (Figure 1f) who was compound heterozygous for p.R683W in one XPD allele and a premature stop codon p.Q452X mutation in the other allele25, 35, 36 (Table 1 and Figure 2). Besides severe sunburning on minimal sun exposure, and extensive freckle-like pigmentation, he had >300 skin cancers documented, including 24 cutaneous melanomas, 284 basal cell carcinomas, and 12 squamous cell carcinomas, despite treatment with oral retinoids.35 He died at the age of 39 years owing to neurological degeneration.
Patient XP34BE was a Caucasian male with a mild XP phenotype (Figure 1g) who had a p.R683W mutation from one XPD allele and a p.I199Pfs*52 mutation from the other allele, which arose from a splice mutation that gave rise to an insertion of eight nucleotides in the mRNA and a predicted truncated protein (Table 1 and Figure 2). He had a sister (XP35BE) who had the same mutations and a similar mild XP phenotype.10, 25 He never developed skin tumors and had no evidence of neurological abnormalities by age 32 years.
Eight of the nine patients had decreased DNA repair as measured by post-UV unscheduled DNA synthesis or post-UV fibroblast survival10, 25 (Table 1). The only exception was patient TTD351BE.
Abnormal CYP24 induction in XPD mutant fibroblasts indicates abnormal transactivation of the VDR
We analyzed by real-time quantitative RT–PCR the expression of two genes known to be under the control of VDR. One of these target genes, CYP24, encodes the cytochrome P450 enzyme vitamin D3 hydroxylase that catalyzes the inactivation of vitamin D (VD). We used normal cells to determine the time and dose–response curves for CYP24 expression. The peak of CYP24 mRNA expression after vitamin D treatment was reached after about 12 h and then did not show a significant change, suggesting that it had reached the expression limit for that concentration of vitamin D (100 nM) (Figure 3a). A similar time course was present in the TTD cells, although at lower peak levels (Supplementary Figure 1). CYP24 expression increased with vitamin D concentration in a dose-dependent manner (Supplementary Figure 2b). The fold increase in CYP24 mRNA of treated relative to untreated control (induction ratio) was in the same range as that described previously for normal fibroblasts39 (Figure 3a and Supplementary Figure 2a). Given the strong CYP24 mRNA expression obtained from incubating the wild-type cells for 12 h with 100 nM vitamin D, we chose to use these same conditions for the patients’ cells to allow comparison.

VDR expression of XPD mutant fibroblasts in XP, TTD, XP/TTD, and normal cells. Relative mRNA expression of the VDR target genes CYP24 and OPN in normal, TTD, XP/TTD, and XP fibroblasts after treatment with VD was measured using qPCR. The values were normalized relative to 18S RNA expression. Each panel represents the results of two independent experiments performed with duplicate samples (*P<0.05; **P<0.01, Student’s t-test compared with normal controls – black bars). (a) Time response control. CYP24 expression in normal (AG04438) fibroblasts incubated for different lengths of time with or without vitamin D (100 nM) was measured. (b) CYP24 induction ratio of treated cells relative to untreated cells for the different cell strains. (c) Time response control. OPN expression in normal (AG04438) fibroblasts incubated for different lengths of time with or without vitamin D (100 nM) was measured. (d) OPN expression of normal, TTD, XP/TTD, and XP fibroblasts after 12 h of incubation with or without vitamin D (100 nM).
The response to vitamin D treatment varied with different cell lines and appears to be influenced by both alleles in these compound heterozygous patients. TTD cells TTD354BE, TTD412BE, TTD404BE, and XPTTD306BE had a significantly (P<0.05) reduced induction ratio when compared with the induction ratio of normal fibroblasts (∼13 000-fold in AG04438 and ∼16 000-fold in AG13145) (Figure 3b). XPTTD306BE had the greatest decrease, with only about a ∼300-fold induction ratio. By comparison, XP29BE and XP34BE had ∼3000- and ∼4000-fold induction ratios, respectively. This reduction is in accord with a previous study of CYP24 mRNA expression.25 Of note, TTD404BE and XP17BE both have the same D681N allele and both have reduced CYP24 induction ratios. Interestingly, these same six TTD, XP/TTD, and XP cell strains also had elevated relative CYP24 baseline levels compared with the normal control (Supplementary Figure 2c, lower panel). In contrast, the TTD and XPTTD cells (TTD351BE, XPTTD306BE) with the same p.R722W mutation in one allele (Figure 2) had opposite responses (Figure 3b and Supplementary Figure 2c). TTD351BE, with an elevated CYP24 response, is the only TTD cell line with normal DNA repair (Table 1 and Boyle et al10). In accord with previous studies,25 these results suggest that the second mutated allele (R378H in TTD351BE and S51F in XPTTD306BE) may also influence the response. Also in agreement with the earlier study,25 XP cells XP17BE, XP29BE, and XP34BE have the same R683W mutation, but differ in their responses to vitamin D (Figure 3b and Supplementary Figure 2c) and have different second mutated alleles.
Abnormalities in OPN induction in XPD mutant fibroblasts further indicate mutation-dependent aberrant transactivation of the VDR
To see whether this effect also involves other VDR target genes, we measured mRNA expression of the OPN gene. Osteopontin is implicated in a variety of physiological functions, including bone remodeling,40 neutrophil chemotaxis,41 and immune regulation.42 OPN expression in normal cells increased with vitamin D concentration in a time-dependent manner (Figure 3c) and in a dose-dependent manner (Supplementary Figure 3b). Similar to our CYP24 data (Figure 3b), we found that the greatest reduction in OPN induction ratio of treated to untreated cells was in TTD354BE and TTD412BE cells and, to a lesser extent, in XP17BE cells (Figure 3d and Supplementary Figure 3c). Considering these similarities of the abnormalities in CYP24 and OPN expression, the data suggest that VDR transactivation may be less responsive to vitamin D stimulation in some of the XPD mutant cells.
We examined ICAM1, a gene that is not known to be responsive to VD, as a negative control. There was no significant increase in the ratio of expression of ICAM1 following VD treatment as compared with untreated cells in normal, or XPD mutant TTD or XP cells (Supplementary Figure 4).
KLF9 expression in XPD mutant fibroblasts
TTD patients often have developmental neurological abnormalities such as microcephaly and brain dysmyelination,1 whereas XP patients may have progressive neurological degeneration.4, 7, 8 The thyroid hormone receptor has been shown to be crucial in normal brain development.43 We measured the expression of KLF9 (also known as BTEB), a thyroid receptor-responsive gene in the fibroblasts.25, 44 The normal cells had a time- and dose-dependent (Supplementary Figures 5a and b) induction of KLF9 by treatment with triiodothyronine (T3). The peak of KLF9 mRNA induction ratio in normal cells after treatment was reached after about 6 h. However, the relative amount of KLF9 mRNA expression from cells with or without T3 treatment increased over time through 72 h. KLF9 induction ratio of treated cells over untreated cells was slightly reduced in TTD354BE, TTD412BE, XPTTD306BE and XP29BE, when compared with normal cells, but these results were not statistically significant (Supplementary Figure 5c).
Discussion
From the group of patients we examined at NIH,8 we selected TTD, XP, and XP/TTD patients with XPD mutations who had markedly different clinical features (Table 1). These patients all were compound heterozygotes for XPD mutations but shared some of the same mutations (Table 1 and Figure 2). TFIIH phosphorylation of nuclear receptors or their co-activated partners (such as Ets1 for VDR) is absent in certain XPD mutants (e.g., p.R683W mutants)24, 45 and intracellular TFIIH concentration is reduced in TTD fibroblasts.10, 16, 23 XPD is a structural bridge between the CAK complex (consisting of cdk7, cycH, and Mat1) and the TFIIH core, which together regulates RNA polymerase II function by phosphorylating its C-terminal domain. This step controls the transition from transcription initiation to elongation.24, 46, 47 Thus, abnormalities in activation of nuclear receptor may be involved in some of the clinical features in these patients.
Ueda et al25 demonstrated that each XPD allele contributes to the CYP24 response. While it is not possible to make unambiguous conclusions from non-isogenic human cell strains, this effect might be mutation dependent and be related to both XPD alleles. TTD351BE and XPTTD306BE cells both had p.R722W mutations, but the ratio of vitamin D stimulated to basal CYP24 expression was elevated in TTD351BE cells, but reduced in XPTTD306BE cells compared with the normal controls (Figure 3b). The second allele in each cell line (p.R378H and p.S51F, respectively) thus might contribute to this differential response. Similar second allele mutation dependency could be evident in the three XP cell lines with p.R683W mutations. This is in agreement with an earlier study25 that reported separate activities of each allele in XP29BE and XP34BE cells. Interestingly, cells with the p.R683W mutation also had a poor response to retinoid receptor stimulation,24 and patient XP29BE, with this mutation, had a poor response to oral retinoid therapy for the prevention of skin cancers despite typical retinoid-related toxicities.35
As reported previously,25 there was no significant reduction in the OPN induction ratios for the XP29BE and XP34BE cells compared with normal cells. However, XP17BE cells and three of the TTD cell strains (TTD354BE, TTD412BE, and TTD351BE) had reduced OPN induction ratios compared with the normal controls (Figure 3d). The results for KLF9, the response gene for the thyroid hormone receptor, were similar, although the magnitude of change was much smaller and would require a larger sample size to elucidate.
We did not observe a correlation between the nuclear receptor transactivation abnormalities and the different clinical phenotypes. This may be related to the fact that we were only able to study skin fibroblasts from our patients after birth. These cells may not be the ones that are most severely affected by this transactivation abnormality. For example, more dramatic effects may be seen in osteocytes for VDR transactivation and CNS glial cells for thyroid receptor transactivation as reported in TTD mice,43 or in fetal cells during embryonic development. Each XPD mutation might affect the transactivation process differently.25 The combination would be expected to lead to variability in nuclear receptor activity among different TTD and XP patients.
References
Faghri S, Tamura D, Kraemer KH, DiGiovanna JJ : Trichothiodystrophy: a systematic review of 112 published cases characterises a wide spectrum of clinical manifestations. J Med Genet 2008; 45: 609–621.
Ruenger TM, DiGiovanna JJ, Kraemer KH : Hereditary diseases of genome instability and DNA repair; in Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, (eds):: Fitzpatrick's Dermatology in General Medicine 7 edn New York: McGraw Hill, 2008, pp 1311–1325.
Nakabayashi K, Amann D, Ren Y et al: Identification of C7orf11 (TTDN1) gene mutations and genetic heterogeneity in nonphotosensitive trichothiodystrophy. Am J Hum Genet 2005; 76: 510–516.
Kraemer KH, Lee MM, Scotto J : Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol 1987; 123: 241–250.
Kraemer KH, Lee MM, Andrews AD, Lambert WC : The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol 1994; 130: 1018–1021.
Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ : Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype–phenotype relationship. Neuroscience 2007; 145: 1388–1396.
DiGiovanna JJ, Kraemer KH : Shining a light on xeroderma pigmentosum. J Invest Dermatol 2012; 132 ((Part 2)): 785–796.
Bradford PT, Goldstein AM, Tamura D et al: Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 2011; 48: 168–176.
Kleijer WJ, Laugel V, Berneburg M et al: Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair 2008; 7: 744–750.
Boyle J, Ueda T, Oh KS et al: Persistence of repair proteins at unrepaired DNA damage distinguishes diseases with ERCC2 (XPD) mutations: cancer-prone xeroderma pigmentosum vs. non-cancer-prone trichothiodystrophy. Hum Mutat 2008; 29: 1194–1208.
Broughton BC, Berneburg M, Fawcett H et al: Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinical outcomes of mutations in the XPD gene. Hum Mol Genet 2001; 10: 2539–2547.
Bergmann E, Egly JM : Trichothiodystrophy, a transcription syndrome. Trends Genet 2001; 17: 279–286.
Egly JM, Coin F : A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair 2011; 10: 714–721.
Hashimoto S, Egly JM : Trichothiodystrophy view from the molecular basis of DNA repair/transcription factor TFIIH. Hum Mol Genet 2009; 18: R224–R230.
Vermeulen W, Bergmann E, Auriol J et al: Sublimiting concentration of TFIIH transcription/DNA repair factor causes TTD-A trichothiodystrophy disorder. Nat Genet 2000; 26: 307–313.
Botta E, Nardo T, Broughton BC, Marinoni S, Lehmann AR, Stefanini M : Analysis of mutations in the XPD gene in Italian patients with trichothiodystrophy: site of mutation correlates with repair deficiency, but gene dosage appears to determine clinical severity. Am J Hum Genet 1998; 63: 1036–1048.
Emmert S, Ueda T, Zumsteg U et al: Strict sun protection results in minimal skin changes in a patient with xeroderma pigmentosum and a novel c.2009delG mutation in XPD (ERCC2). Exp Dermatol 2009; 18: 64–68.
Fan L, Fuss JO, Cheng QJ et al: XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell 2008; 133: 789–800.
Kobayashi T, Uchiyama M, Fukuro S, Tanaka K : Mutations in the XPD gene in xeroderma pigmentosum group D cell strains: confirmation of genotype-phenotype correlation. Am J Med Genet 2002; 110: 248–252.
Taylor EM, Broughton BC, Botta E et al: Xeroderma pigmentosum and trichothiodystrophy are associated with different mutations in the XPD (ERCC2) repair/transcription gene. Proc Natl Acad Sci USA 1997; 94: 8658–8663.
Dilworth FJ, Chambon P : Nuclear receptors coordinate the activities of chromatin remodeling complexes and coactivators to facilitate initiation of transcription. Oncogene 2001; 20: 3047–3054.
Glass CK, Rosenfeld MG : The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 2000; 14: 121–141.
Dubaele S, Proietti dS, Bienstock RJ et al: Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell 2003; 11: 1635–1646.
Keriel A, Stary A, Sarasin A, Rochette-Egly C, Egly JM : XPD mutations prevent TFIIH-dependent transactivation by nuclear receptors and phosphorylation of RARalpha. Cell 2002; 109: 125–135.
Ueda T, Compe E, Catez P, Kraemer KH, Egly JM : Both XPD alleles contribute to the phenotype of compound heterozygote xeroderma pigmentosum patients. J Exp Med 2009; 206: 3031–3046.
Moslehi R, Signore C, Tamura D et al: Adverse effects of trichothiodystrophy DNA repair and transcription gene disorder on human fetal development. Clin Genet 2010; 77: 365–373.
Tamura D, Merideth M, DiGiovanna JJ et al: High-risk pregnancy and neonatal complications in the DNA repair and transcription disorder trichiothiodystrophy: report of 27 affected pregnancies. Prenat Diagn 2011; 31: 1046–1053.
Robbins JH, Kraemer KH, Lutzner MA, Festoff BW, Coon HG : Xeroderma pigmentosum. An inherited disease with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann Intern Med 1974; 80: 221–248.
Khan SG, Muniz-Medina V, Shahlavi T, Baker CC, Inui H, Ueda T et al: The human XPC DNA repair gene: arrangement, splice site information content and influence of a single nucleotide polymorphism in a splice acceptor site on alternative splicing and function. Nucleic Acids Res 2002; 30: 3624–3631.
Zhou X, Khan SG, Tamura D et al: Brittle hair, developmental delay, neurologic abnormalities, and photosensitivity in a 4-year-old girl. J Am Acad Dermatol 2010; 63: 323–328.
Viprakasit V, Gibbons RJ, Broughton BC et al: Mutations in the general transcription factor TFIIH result in beta- thalassaemia in individuals with trichothiodystrophy. Hum Mol Genet 2001; 10: 2797–2802.
Takayama K, Danks DM, Salazar EP, Cleaver JE, Weber CA : DNA repair characteristics and mutations in the ERCC2 DNA repair and transcription gene in a trichothiodystrophy patient. Hum Mutat 1997; 9: 519–525.
De Boer J, De Wit J, Van Steeg H et al: A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol Cell 1998; 1: 981–990.
Graham JM, Anyane-Yeboa K, Raams A et al: Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am J Hum Genet 2001; 69: 291–300.
Kraemer KH, DiGiovanna JJ, Moshell AN, Tarone RE, Peck GL : Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N Engl J Med 1988; 318: 1633–1637.
Wang Y, DiGiovanna JJ, Stern JB et al: Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci USA 2009; 106: 6279–6284.
Liang C, Kraemer KH, Morris A, Schiffmann R, Price VH, Menefee E et al: Characterization of tiger-tail banding and hair shaft abnormalities in trichothiodystrophy. J Am.Acad.Dermatol 2005; 52: 224–232.
Andrews AD, Barrett SF, Robbins JH : Xeroderma pigmentosum neurological abnormalities correlate with colony-forming ability after ultraviolet radiation. Proc.Natl.Acad.Sci.U.S.A 1978; 75: 1984–1988.
Tashiro K, Abe T, Oue N, Yasui W, Ryoji M : Characterization of vitamin D-mediated induction of the CYP 24 transcription. Mol Cell Endocrinol 2004; 226: 27–32.
Reinholt FP, Hultenby K, Oldberg A, Heinegard D : Osteopontin – a possible anchor of osteoclasts to bone. Proc Natl Acad Sci USA 1990; 87: 4473–4475.
Koh A, da Silva AP, Bansal AK et al: Role of osteopontin in neutrophil function. Immunology 2007; 122: 466–475.
Wang KX, Denhardt DT : Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev 2008; 19: 333–345.
Compe E, Malerba M, Soler L, Marescaux J, Borrelli E, Egly JM : Neurological defects in trichothiodystrophy reveal a coactivator function of TFIIH. Nat Neurosci 2007; 10: 1414–1422.
Moeller LC, Dumitrescu AM, Walker RL, Meltzer PS, Refetoff S : Thyroid hormone responsive genes in cultured human fibroblasts. J Clin Endocrinol Metab 2005; 90: 936–943.
Drane P, Compe E, Catez P, Chymkowitch P, Egly JM : Selective regulation of vitamin D receptor-responsive genes by TFIIH. Mol Cell 2004; 16: 187–197.
Chen D, Riedl T, Washbrook E et al: Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol Cell 2000; 6: 127–137.
Fuss JO, Tainer JA : XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair (Amst) 2011; 10: 697–713.
Acknowledgements
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (NIH) and by the European Research Council (ERC-2008-TRANSREACT) (authors EC and J-ME). Support for author Zhou was made possible through the Clinical Research Training Program, a public–private partnership supported jointly by the NIH and Pfizer Inc. (via a grant to the Foundation for NIH from Pfizer Inc.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors state no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Zhou, X., Khan, S., Tamura, D. et al. Abnormal XPD-induced nuclear receptor transactivation in DNA repair disorders: trichothiodystrophy and xeroderma pigmentosum. Eur J Hum Genet 21, 831–837 (2013). https://doi.org/10.1038/ejhg.2012.246
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.246
Keywords
This article is cited by
-
Facial clues to the photosensitive trichothiodystrophy phenotype in childhood
Journal of Human Genetics (2023)
-
Differences in peripheral neuropathy in xeroderma pigmentosum complementation groups A and D as evaluated by nerve conduction studies
BMC Neurology (2021)
-
Mutations in the TTDN1 Gene Are Associated with a Distinct Trichothiodystrophy Phenotype
Journal of Investigative Dermatology (2015)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}