
Abstract
The most common form of Dopa-responsive dystonia (DRD) is caused by heterozygous mutations in the GTP cyclohydrolase I (GCH1) gene. We screened two unrelated, DRD-symptomatic Chinese Han individuals, for GCH1 gene mutations by direct sequencing. As the clinical manifestations of DRD are highly variable, we also explored the association between genotype and phenotype in all Chinese DRD patients reported so far in the literature, comprising 62 DRD-affected patients from 36 Chinese families. Two novel missense mutations (T94M, L145F) and a novel variant (c. 453+6 G>T) were identified in our two new patients. None of these variants was detected in 200 healthy controls. On the basis of this and other reports, heterozygous mutations were detected in 90.3% of Chinese Han subjects with DRD. Seeming the age of onset for males and females, the mean age was 13 years older in males than in females (P=0.006). Different mutation types did not show any significant differences in age of onset, gender composition, initial symptoms, or the L-dopa dose that abolished the symptoms. Among DRD patients lacking missense or exon–intron boundary mutations, 68.4% were found to possess a large deletion in GCH1, which were detected by multiplex ligation-dependent probe amplification. Most GCH1 mutations were found to cluster in two regions of the coding sequence, suggesting the probable existence of mutation hotspot for the first time. The genotype–phenotype correlation described here may improve our understanding of DRD in Chinese individuals.
Similar content being viewed by others

Introduction
Dopa-responsive dystonia (DRD), first described by Segawa et al1, is a rare inherited movement disorder with a prevalence of 0.5–1 per million.2 It is characterized by fluctuating dystonia that develops during childhood and that features postural dystonia of the lower limbs, which becomes aggravated toward evening and alleviates after sleep.3 Arm dystonia, hyperreflexia, and parkinsonism (bradykinesia, hypomimia, postural instability) are also common features.4 The hallmark feature of all clinical subtypes of DRD is a dramatic and sustained response to low-dose levodopa treatment.3
DRD has two modes of inheritance: the commonest autosomal-dominant form is caused by heterozygous mutation in the GTP cyclohydrolase 1 (GCH1, DYT5) gene and more rarely in the sepiaptein reductase (SPR) gene. The autosomal-recessive form is caused by homozygous or compound heterozygous mutations of the tyrosine hydroxylase gene (TH).5, 6 GTP cyclohydrolase 1 (GTPCH1), encoded by GCH1, is a rate-limiting enzyme, that catalyzes the first step in the biosynthesis of tetrahydrobiopterin (BH4). BH4 is an essential cofactor for tyrosine hydroxylase (TH), tryptophan hydroxylase, and phenylalanine hydroxylase. Up to now, >100 GCH1 mutations have been reported among different ethnic populations (HGMD, http://www.hgmd.org).
DRD is clinically and genetically heterogeneous. Clinical manifestations of DRD differ among families, and even among patients from the same family.9 However, the clinical and genetic heterogeneity associated with the disease means that large number of DRD patients are needed in order to examine the correlations between phenotype and genotype, most studies to date have been conducted on relatively limited patient populations.
Most reported cases of DRD have come from Japan and South Asia, with few analyses of Chinese DRD patients published.9, 10, 11, 12, 13 To extent these studies to China and increase the clinical value of phenotype–genotype analysis of DRD patients, in the present study, we screened GCH1 mutations in two families containing members newly diagnosed with DRD. We also conducted phenotype–genotype correlation analysis on the largest cohort of Chinese DRD patients to date, comprising all patients previously reported.
Materials and methods
GCH1 screening
Our study included two patients with sporadic DRD and four clinically unaffected relatives from two families, as well as 200 healthy controls. All subjects belonged to the Chinese Han ethnic group. All patients fulfilled established criteria for DRD.14 Cranial magnetic resonance imaging revealed no abnormalities. The symptoms of these patients were completely eliminated by the administration of a tablet containing low-dosage L-dopa and benserazide (50–60 mg/d). This study was approved by the local ethics committee, and written informed consent was obtained from all subjects.
Six primer pairs targeting the GCH1 gene were designed using Primer 3.0 (http://frodo.wi.mit.edu/primer3/). The entire GCH1 coding region, as well as the exon–intron boundary were amplified by PCR. Direct sequencing was performed using an automated DNA sequencer (ABI 3730, Applied Biosystems, CA, USA). Sample sequences were compared with the GCH1 genomic (Ensembl Gene ID: ENSG00000131979). The large exonic deletion of GCH1 was detected by multiplex ligation-dependent probe amplication analysis.
ClustalW2 (http://www.ebi.ac.uk/clustalw/) was used to analyze phylogenetic conservation of the mutation sites by aligning amino-acid sequences from several species retrieved from the Ensembl Genome Browser. The effect of the observed intron variant on GCH1 mRNA splicing was assessed using the electronic tool NNSplice (http://www.fruitfly.org/seq_tools/splice.html). The functional effects of missense mutations in GTPCH1 were predicted using Polyphen (http://genetics.bwh.harvard.edu/pph/), SNAP (http://cubic.bioc.columbia.edu/services/SNAP/), and PMut programs (http://mmb2.pcb.ub.es:8080/PMut/).
Genotype–phenotype correlation
Genotype–phenotype correlation analysis was carried out using clinical and genetic information from all Chinese DRD patients, described so far in the literature, including the two DRD patients in the present cohort. Thus the analysis involved a total of 62 DRD patients from 36 families, including 25 sporadic DRD cases and 37 family DRD cases.9, 10, 11, 12, 13 All patients in this analysis met the diagnosis criteria for DRD.14 Each mutation was classified as a (1) missense mutation; (2) exon–intron boundary variant, that is, a variant located in an exon–intron boundary region; (3) large deletion,which can be deleted only by multiple ligation-dependent probe amplification analysis; or (4) small deletion, which can be detected only by direct sequencing.
Statistical analysis
Descriptive statistics, including frequency counts and means, were used to describe the group of patients with DRD. The χ2-test and one-way ANOVA were used to test for significant differences between groups. The independent-samples t-test was used to assess differences in age of onset between groups. A two-tailed P-value <0.05 was considered significant. All statistical analyses were performed using the Statistical Package for Social Sciences for Windows (SPSS, version 18.0; SPSS Inc., Chicago, IL, USA).
Results
Novel mutations
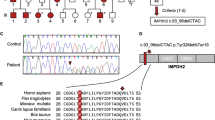
Two novel heterozygous mutations and a novel exon–intron boundary variant were detected in our study. The novel mutation T94M (c. 281C>T) was found in case A, which involves a transition from polar threonine to nonpolar methionine. The other novel mutation, L145F (c. 435 G>T), and the novel exon–intron boundary variant, c. 453+6 G>T, were both found in case B (Figure 1a). We tested the asymptomatic parents of both patients, and found that case A harbored the same mutation as her daughter, while neither variant was found in the parents of case B. None of the three variants was found in 200 healthy Chinese controls. Both the T94M and L145F mutations occur in regions highly conserved across several species (Figure 1b). Both appear to be pathogenetic, as predicted by Polyphen, PMut, and SNAP software. The variant c.453+6 G>T probably also produces a functional change in the GTPCH1 protein, as NNSplice predicts that it increases the acceptor score of the splice site by 0.17. The clinical manifestations of the patients and the results of the prediction software are summarized in Table 1.

(a) DNA sequence chromatograms of GCH1 regions showing the heterzygous substitutions C>T (T94M) in exon 1, G>T (L145F) in exon 2, and G>T (c. 453+6 G>T) at an exon–intron boundary. The electropherogram of the sense strand is shown at the top and a limited reading frame depicting the corresponding amino-acid substitutions is shown below. (b) Partial amino-acid sequence alignment of GTPCH1 proteins from various vertebrate species.
Clinical descriptions and genotypes of Chinese DRD patients
The clinical features of the 62 patients included in our study are listed in Table 2. Females strongly outnumbered males by 52–10. The mean age of onset was 13.9±14.0 years for the entire group, 11.6±11.5 years in females, and 24.6±20.2 years in males (P=0.006). The age of onset was about 13 years earlier for females than for males. Of all patients, 55 (88.7%) initially presented with foot or leg dystonia. The remaining seven patients (11.3%) showed parkinsonism as an onset symptom. The age of onset differed significantly between DRD patients with dystonia at onset (10.2±7.0 years) and those with parkinsonism at onset (42.0±21.6 years, P<0.001). The mean delay between onset and diagnosis was 12.8±10.4 years. The median L-dopa dose that abolished DRD symptoms was 133.8±94.53 mg/d.
Of the 62 DRD patients, 56 (90.7%) possessed mutations in GCH1, including missense mutation, a small deletion, or large deletion, or an exon–intron boundary variant. This proportion is much higher than the 50%–87% observed among non-Chinese ethinicities’.7, 8, 15 The proportion of sporadic DRD patients with GCH1 variants was 84.0%, compared with 94.6% for family DRD patients (P=0.21). The large exonic deletion of GCH1 was present in 13 of 62 Chinese patients (20.1%), much higher than the 8.0% reported in a European population.16 In fact, this large deletion occurred in 13 of 19 (68.4%) mutation-negative DRD patients, who lacked other GCH1 mutation determined by gene sequencing.
Genotype–phenotype correlation
Phenotypic details of DRD patients and mutations are presented in Table 2. GCH1 mutations were present at significantly different proportions among Chinese DRD patients, missense 62.5%; small deletion, 8.1%; large deletion, 20.1%; and exon–intron boundary variations, 4.8% (P<0.001). Age of DRD onset and gender composition did not differ significantly among these groups (P=0.638, and P=0.051, respectively). Similarly, the groups did not differ significantly in initial symptoms, initial location, diurnal fluctuation, or effective L-dopa dose (P>0.05). The large deletion group also needed a higher effective L-dopa dose than did the other groups.
Discussion
This study describes two novel mutations and a novel intron variant in GCH1 in two Chinese DRD patients, thereby expanding the spectrum of mutations associated with DRD. The mutation T94M, which involves a change from a polar threonine to a nonpolar methionine, was found not only in case A, but also in her asymptomatic mother. The fact that the mother was asymptomatic may reflect the incomplete penetrance/expression of GCH1 gene mutations.17 Interestingly, both the mutation c L145F and the variant c. 453+6G>T were identified in case B, but not in her parent. These are probably ‘de novo’ variants, which are common in Chinese DRD patients.12
The missense mutations T94M and L145F are highly pathogenetic, as they affect evolutionarily conserved residues in GTPCH1 and were absent from our 200 controls. Both are predicted to harm protein function by PMut, SNAP, and Polyphen. The variant c. 453+6 G>T is located in the exon–intron boundary region, not far from the position of L145F. The exon–intron boundaries are special regions that usually have important functional roles in proteins.10 Indeed, NNSplice predicts that variant c. 453+6 G>T affects the acceptor score of the splice site. We speculate that L145F and c. 453+6G>T interact synergistically in the pathogenesis of case B.
Both patients in our study presented typical DRD symptoms: early onset, at a mean age of 5.5 years; dystonia in the lower legs; diurnal fluctuations; and excellent response to low-dose levodopa. Diagnosis of DRD is difficult as clinical features are highly variable, ranging from focal to generalized dystonia, parkinsonism or only subtle signs.3, 7, 18 As a result, misdiagnosis and delayed treatment are common. When we combined our two new DRD patients with the 60 Chinese patients, described so far in the literature, we found the mean delay between onset and diagnosis to be 12.8±10.4 year. This probably reflects a lack of public awareness about the disease, as well as deficiencies in the regional healthcare system for diagnosing and treating it. Levodopa is a highly effective treatment for DRD, with most cases showing complete remission if treatment is administered early. However, delays or errors in the treatment can aggravate symptoms and seriously affect patients’ quality of life, even causing irreversible damage like scoliosis. Therefore, genetic analysis of genes associated with DRD cases may be an effective method for confirming the diagnosis, guiding treatment, and informing genetic counseling.
In this study, the natural history of DRD and genotype–phenotype correlation were analyzed in the largest number of Chinese patients to date. The proportion of Chinese DRD patients with GCH1 mutations is up to 90.3%, which is much higher than the 50%–87% reported previously.7, 8 The rate of the large GCH1 deletion was very high in Chinese DRD patients (20.1%), especially in the mutation-negative ones testing by gene sequencing (68.4%). Furthermore, the proportion 20.1% is much higher than the 8.0% reported in a European population,16 which suggests the need to test for the large deletion by multiplex ligation-dependent probe amplification in Chinese DRD patients. The genotype–phenotype correlation analysis did not provide much new information beyond what has been published, probably because the type of mutation and clinical phenotype do not show a strong association in Chinese DRD patients. The other possibility is that an association exists but the statistical power of our study was too small to detect it, given the relatively low number of patients with large and small deletions in our sample.
We found that most mutations in Chinese DRD patients clustered in two regions of the GCH1 coding sequence, (base pair range between 210–360 and 550–650) (Figure 2). These regions may be mutation hotspots, which have not previously been suggested for DRD pathogenesis. However, a founder effect cannot be excluded as a possible explanation. A missense mutation in GCH1 leading to the amino-acid substitution, G203R was found to occur in 10 of 56 (17.9%) Chinese DRD patients in our study, suggesting that the founder effect does occur in this patient population. Determining whether the concentration of mutations indicates hotspots in GCH1 will require studies with larger samples and multi-ethnic genotype–phenotype correlation analysis.

Age of onset of DRD was not associated with mutation position (n=40 with DRD). The number of mutations was probably associated with mutation position (n=40 with DRD). With most mutations clustering in two regions of coding sequence: base pair ranges between 210–360 and 550–650.
The results of our study suggest that analysis based on clinical manifestations and genetic testing may be effective for diagnosing and treating DRD, and genotype–phenotype correlation may help physicians to evaluate risk in DRD families and provide appropriate counseling. Genetic variants that may contribute to pathogenesis include not only missense mutations, but also variants in special regions such as intron–exon boundaries, which therefore deserve more attention. Prediction software may help us understand such variants in greater detail, but functional studies are ultimately needed to test their role in the disease.
References
Segawa M, Hosaka A, Miyagawa F, Nomura Y, Imai H : Hereditary progressive dystonia with marked diurnal fluctuation. Adv Neurol 1976; 114: 215–233.
Segawa M, Nomura Y, Nishiyama N : Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol 2003; 54: S32–S45.
Nygaard TG, Marsden CD, Duvoisin RC : Dopa-responsive dystonia. Adv Neurol 1988; 50: 377–384.
Nygaard TG, Takahashi H, Heiman GA, Snow BJ, Fahn S, Calne DB : Long-term treatment response and fluorodopa positron emission tomographic scanning of parkinsonism in a family with dopa-responsive dystonia. Ann Neurol 1992; 32: 603–608.
Asmus F, Gasser T : Dystonia-plus syndromes. Eur J Neurol 2010; 17: 37–45.
Steinberger D, Blau N, Goriuonov D et al: Heterozygous mutation in 5’-untranslated region of sepiapterin reductase gene (SPR) in a patient with dopa-responsive dystonia. Neurogenetics 2004; 5: 187–190.
Bandmann O, Valente EM, Holmans P et al: Dopa-responsive dystonia: a clinical and molecular genetic study. Ann Neurol 1998; 44: 649–656.
Hagenah J, Saunders-Pullman R, Hedrich K et al: High mutation rate in dopa-responsive dystonia: detection with comprehensive GCH1 screening. Neurology 2005; 64: 908–911.
Wu-Chou YH, Yeh TH, Wang CY et al: High frequency of multiexonic deletion of the GCH1 gene in a Taiwanese cohort of dopa-response dystonia. Am J Med Genet Neuropsychiatr Genet 2010; 153B: 903–908.
Cao L, Zheng L, Tang WG et al: Four novel mutations in the GCH1 gene of Chinese patients with dopa-responsive dystonia. Mov Disord 2010; 25: 755–760.
Liu X, Zhang SS, Fang DF et al: GCH1 mutation and clinical study of Chinese patients with dopa-responsive dystonia. Mov Disord 2010; 25: 447–451.
Hu FY, Xu YM, Yu LH, Ma MY, He XH, Zhou D : A novel missense mutation in GTP cyclohydrolase I (GCH1) gene causes dopa-responsive dystonia in Chinese Han population. Eur J Neurol 2011; 18: 362–364.
Wu ZY, Lin Y, Chen WJ et al: Molecular analyses of GCH-1, TH and parkin genes in Chinese dopa-responsive dystonia families. Clin Genet 2008; 74: 513–521.
Abeling NG, Duran M, Bakker HD et al: Sepiapterin reductase deficiency an autosomal recessive DOPA-responsive dystonia. Mol Genet Metab 2006; 89: 116–120.
Steinberger D, Korinthenberg R, Topka H et al: Dopa-responsive dystonia: mutation analysis of GCH1 and analysis of therapeutic doses of L-dopa. Neurology 2000; 55: 1735–1737.
Zirn B, Steinberger D, Troidl C et al: Frequency of GCH1 deletions in Dopa-responsive dystonia. J Neurol Neurosurg Psychiatry 2008; 79: 183–186.
Furukawa Y : Update on dopa-responsive dystonia: locus heterogeneity and biochemical features. Adv Neurol 2004; 94: 127–138.
Steinberger D, Topka H, Fischer D, Müller U : GCH1 mutation in a patient with adult-onset oromandibular dystonia. Neurology 1999; 52: 877–879.
Acknowledgements
This study was supported by a Sichuan Key Project of Science and Technology grant (No. 2010SZ0086) and the National Natural Science Foundation of China (Number 30700243). We thank all the subjects who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Yu, L., Zhou, H., Hu, F. et al. Two novel mutations of the GTP cyclohydrolase 1 gene and genotype–phenotype correlation in Chinese Dopa-responsive dystonia patients. Eur J Hum Genet 21, 731–735 (2013). https://doi.org/10.1038/ejhg.2012.239
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.239
