
Abstract
Familial porencephaly, leukoencephalopathy and small-vessel disease belong to the spectrum of disorders ascribed to dominant mutations in the gene encoding for type IV collagen alpha-1 (COL4A1). Mice harbouring mutations in either Col4a1 or Col4a2 suffer from porencephaly, hydrocephalus, cerebral and ocular bleeding and developmental defects. We observed porencephaly and white matter lesions in members from two families that lack COL4A1 mutations. We hypothesized that COL4A2 mutations confer genetic predisposition to porencephaly, therefore we sequenced COL4A2 in the family members and characterized clinical, neuroradiological and biochemical phenotypes. Genomic sequencing of COL4A2 identified the heterozygous missense G1389R in exon 44 in one family and the c.3206delC change in exon 34 leading to frame shift and premature stop, in the second family. Fragmentation and duplication of epidermal basement membranes were observed by electron microscopy in a c.3206delC patient skin biopsy, consistent with abnormal collagen IV network. Collagen chain accumulation and endoplasmic reticulum (ER) stress have been proposed as cellular mechanism in COL4A1 mutations. In COL4A2 3206delC fibroblasts we detected increased rates of apoptosis and no signs of ER stress. Mutation phenotypes varied, including porencephaly, white matter lesions, cerebellar and optic nerve hypoplasia and unruptured carotid aneurysm. In the second family however, we found evidence for additional factors contributing to the phenotype. We conclude that dominant COL4A2 mutations are a novel major risk factor for familial cerebrovascular disease, including porencephaly and small-vessel disease with reduced penetrance and variable phenotype, which might also be modified by other contributing factors.
Similar content being viewed by others


Introduction
Porencephaly is defined as a cavity in the cerebral parenchyma communicating with the lateral ventricles, which results from destruction of the parenchyma after a developmental defect or an intracerebral haemorrhage. Porencephaly has both genetic and non-genetic causes. Mutations in the type IV collagen alpha-1 (COL4A1) gene, coding for the procollagen alpha-1 chain of collagen IV, have been associated with variable phenotypes. COL4A1 mutations predispose to germinal matrix haemorrhage in late pregnancy or around birth, which can lead to deep venous infarction with subsequent tissue necrosis and porencephalic cavitation. When the haemorrhage occurs during gestation or around birth it usually manifests as congenital hemiplegia.1, 2, 3, 4, 5 Familial porencephaly with congenital hemiplegia has been ascribed to autosomal dominantly inherited mutations in the COL4A1 gene. Although a number of rare monogenetic disorders may predispose to intracerebral haemorrhage, such as CADASIL, Fabry's disease, autosomal dominant amyloid angiopathy, MELAS, homocystinuria, Ehlers-Danlos syndrome type IV, Marfan and Maeda syndrome,1, 2, 5 only mutations of COL4A1 have been associated with familial porencephaly.3 The phenotypic manifestation of COL4A1 mutations is, however, quite variable both within and among families, with different presentations in children or adults. As well as porencephaly, COL4A1 mutations predispose to a variety of vascular lesions in the brain and other organs, leading to the conclusion that COL4A1 mutation causes a multisystem disease.6 The cerebral lesions vary from deep intracerebral haemorrhage, to diffuse leukoencephalopathy, or small-vessel disease with deep lacunar infarcts, dilated perivascular spaces, silent microbleeds or (unruptured) intracranial aneurysms. In mutation carriers, symptoms are variable, including congenital hemiplegia, transient ischaemic attack, adult-onset haemorrhagic stroke, rarely brain infarction, and may even be absent.7, 8, 9, 10 At the severe end of the spectrum, massive prenatal bleeding leading to apparent hydranencephaly, renal agenesis and early demise have been described.11 Other organs can also be affected. Ocular features can be associated with COL4A1 mutations such as retinal arteriolar tortuosity, ocular cataract, anterior segment dysgenesis and microcornea.9, 12, 13, 14, 15 Other manifestations are cardiac arrhythmia, renal cysts, haematuria, hepatic cysts, muscle cramps and elevated creatine kinases. The HANAC syndrome (hereditary angiopathy, nephropathy, aneurysms and muscle cramps) has been specifically associated with mutations in exons 24 and 25 of the COL4A1 gene.8, 16 COL4A1 mutations are often described within a familial context, but can also occur de novo, without a family history of bleeding.11, 17 Although heterogeneous, the common association of the symptoms has led to the definition of general criteria for consideration of COL4A1 genetic testing.6 The identification of a familial COL4A1 mutation helps to monitor complications and supports genetic counselling.
Non-fibrillary collagen IV is a major component of the extracellular matrix and basement membranes and consists of six alpha chains, encoded by distinct and tandem located genes, respectively, on chromosome 13q (COL4A1 and COL4A2), 2q (COL4A3 and COL4A4) and Xq (COL4A5 and COL4A6). COL4A3 to COL4A6 mutations all cause Alport syndrome and benign familial haematuria. COL4A1 and COL4A2 are under regulation of the same promoter and encode for the procollagen IV alpha-1 and IV alpha-2 chains, which assemble to form a heterotrimeric helix with a constant 2:1 ratio, [alpha1(IV)]2[alpha2(IV)]. Among collagen IV genes, only COL4A1 and COL4A2 are ubiquitously expressed.18 This explains why the basement membranes of several epithelia and endothelia can be affected by a COL4A1 mutation. Mutations in the human COL4A1 gene were originally identified after description of the prenatal cerebral and ocular bleeding in mice bearing mutations in the homologue Col4a1 gene.3 Because of the common function, mutations in the collagen IV alpha-2 gene were supposed to cause similar manifestations and in fact, mice bearing heterozygous Col4a2 missense mutations survive the postnatal period but show haemorrhage in the eye, brain, and skin and developmental defects of the eye and brain19 similar to Col4a1 mutants. We observed porencephaly, white matter lesions suggestive of small-vessel disease and aneurysms in members of two families, fulfilling the criteria for COL4A1 testing, but without evidence for COL4A1 mutation. We therefore explored by a candidate gene approach the involvement of COL4A2 and its potential role in human disease.
Subjects and methods
Patient study
All tested individuals and legal caretakers gave their informed consent for the study, according to the Dutch local ethical committee requirements. Magnetic resonance imaging (MRI) and analysis of diffusion tensor imaging data were performed according to Lobel et al.20, 21, 22 Transmission electron microscopy of the skin biopsies was performed in double-blind experiments and independently scored by two investigators.8
Genomic COL4A1 sequence analysis was according to Breedveld et al.7 Primers were designed and analysis performed for the genomic sequence of COL4A2. RT-PCR was performed as described.21 RT-qPCR was performed using KAPA sybr fast qPCR mix ABI prism (KAPA Biosystems, Boston, MA, USA) and the CFX96 Real Time System (Bio-Rad, Veenendaal, The Netherlands). Skin fibroblast culture and test for endoplasmic reticulum (ER) stress was conducted by western blot and RT-qPCR according to Lin et al.23 Staining for the collagen-specific chaperon HSP47 and the ER stress marker KDEL24 was performed under standard culture condition and after stress induced by DTT in control and patient fibroblasts. A fluorescent kit for multiple active caspases was used (Flica apoptosis detection kit, Immunochemistry Technologies, LLC, Bloomington, IN, USA). For additional details see the Supplementary data.
Results
Patient description
Clinical details are summarized in Table 1.
Family A
The proband III-2 was the first of this Caucasian family to present at the age of 6 months with a right-sided hemiparesis (Figure 1a). Her prenatal course was complicated by oligohydramnion. Brain MRI at the age of 3 years and 8 months showed left-sided porencephaly and right-sided periventricular white matter hyperintense lesions on T2 weighted images. At the age of 4.5 years she has a moderate learning disability. Eye fundoscopy is normal. (Figures 2a and b).

Pedigrees of families A and B. Different symbols indicate individuals affected with cerebral vascular disease, while the genotype is indicated under the symbol. The genotype of the tested individuals is indicated: (a) 4165G/G=wild-type sequence; 4165G/A=heterozygous mutation; (b) 3206C/C=wild-type sequence; 3206C/delC=heterozygous mutation. Electroferograms indicate the heterozygous mutation in the COL4A2 sequence of family A c.4165G>A (p.G1389R) (c) and family B c.3206delC leading to frame shift (d). Quantitative, real time PCR analysis showing relative expression of COL4A1 and COL4A2 mRNA in fibroblasts from patient II.2 (family B) with c.3206delC mutation (A2 patient) and a patient bearing a pathogenic c.3321G>C (p.G1067A) mutation in exon 38 of COL4A1 (A1 patient), compared with a control cell line. Results are average of two separate experiments. For each cell line the highest expression is set as 100% on the Y axis (e). Position of the mutations within the schematic representation of the COL4A2 genomic organization (f).

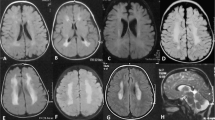
Brain MRI of patients with COL4A2 mutations. (a, b) Axial and coronal T2 of patient III.2 from family A at the age of 2 years indicate ex-vacuo dilation of the left lateral ventricle (porencephaly) (open arrow) and periventricular white matter lesions (solid arrows) resulting from presumed perinatal stroke. (c) FLAIR image illustrates T2 prolongation in the white matter – in patient III.1 from family A at the age of 8 years, suggesting gliosis (arrow). (d) MR angiography of their mother (subject II.1, family A) at adult age shows bilateral internal carotid aneurysms (at the level of cavernous sinus). (e) Axial T2 weighted images of Patient II.2, family B, at the age of 15 years show a porencephalic dilatation of the left occipital ventricle. (f, g) Axial T2 weighted and coronal FLAIR images of patient II.5 from family B at the age of 5 months show porencephaly of the right ventricle with hypoplastic left cerebellar hemisphere. (h–m) Reconstruction of the white matter tracts obtained from magnetic resonance diffusion tensor imaging (MR-DTI) data. (k,l) Patient II-2 (family B) reveals reduced fractional anisotropy in the left radiation optica and tractus corticospinalis at the side of the porencephalic lesion (encircled), compared with an age-matched control (h,i). A restriction of the total ADC was observed in the whole cerebral white matter of the patient (m), compared with an age-matched control (j).
Subject III-1 was born at 38 weeks of a normal gestation and developed normally. A brain MRI at the age of 8 years revealed patchy white matter lesions. Eye fundoscopy is normal (Figure 2c).
The mother (II-2) is asymptomatic at the age of 31 years. The brain MRI was normal, while MRA revealed bilateral internal carotid artery aneurysms at the level of the cavernous sinus (Figure 2d).
Subjects II-4, II-5 and I-1 are all reported to have a right-sided hemiparesis. No medical records are available.
Family B
This consanguineous family of Afghani ancestry includes two probands (II-2 and II-5) (Figure 1b).
Proband II-2 was born at term of a normal gestation with a birth weight of 2500 g (−3 SD). At the age of six months a right-sided hemiparesis was noted. At the age of 16 years he has an intelligence quotient of 48, a dystrophic appearance with microcephaly and a right-sided dystonic paresis. Brain MRI revealed left-sided cerebral atrophy with porencephaly and Wallerian degeneration of the brain stem (Figures 2e, k–m). Ophthalmological examination showed severe myopia with bilateral amblyopia and small optic discs, but no arteriolar tortuosity. Proband II-5's prenatal course was complicated by maternal diabetes and intrauterine growth retardation. A caesarian section was performed at 31 weeks of gestation because of fetal distress. Cerebral ultrasound at birth showed a right-sided subependymal bleeding with venous infarction. At 6 months brain MRI revealed a porencephalic cyst and hypoplastic left cerebellar hemisphere (Figures 2f and g). Physical examination at the age of 1 year showed a left-sided hemiparesis and borderline microcephaly (−2 SD). Ophthalmological examination, renal ultrasound and echocardiogram were normal. Screening for coagulopathies in the neonatal period showed a suspected protein S deficiency, but no analysis was repeated afterwards.
The mother (I-2) has hypothyroidism. She was small at birth and had feeding difficulties. Ophthalmological examination showed high myopia, astigmatism and tilted optic discs with some peripapillary atrophy. Permission for brain imaging was denied. She also delivered two daughters who died of complicated congenital hydrocephalus (subjects II-1 and II-4). Brain MRI of one daughter (II-4) at the age of 2 months showed extreme hydrocephalus with periventricular cystic lesions, stenosis of the Sylvius aqueduct and periventricular, subcortical and cerebellar calcifications (Supplementary Figure S1). Another son (subject II-3) is mildly intellectually disabled with a total intelligence quotient of 62, a normal head circumference and normal brain MRI. Her last pregnancy was uneventful, was monitored by ultrasound and ended with the birth of a third daughter (subject II-6) in the 38th week. At birth cerebral ultrasound revealed a small intraparenchymal bleeding. The girl developed a mild hemiparesis, milder than the probands; her brain MRI revealed a small right-sided cerebral intraparenchymal cyst (Supplementary Figure S2). Extensive coagulopathy screening at the age of 2 years revealed a heterozygote protein S deficiency.
Sequence analysis and expression data
COL4A1
DNA sequence of the 52 coding exons and intron–exon boundaries of COL4A1 in subjects III-1 and III-2 of family A and subjects II-5 and I-1 of family B was normal.7 A maximum number of 13 heterozygote SNPs were observed in the sequence of each individual, which excludes the presence of large deletions in one of the COL4A1 alleles.
COL4A2
Family A: DNA sequencing of part of the 5′ UTR and the 48 exons and intron-exon boundaries of COL4A2 was performed in subjects III-1, III-2, III-3 and II-2. In patient III-2, III-1 and II-2 a heterozygous missense change, c.4165G>A in exon 44, leading to an amino acid substitution of glycine by arginine at position 1389 (p.G1389R) was found (Figure 1c). COL4A2 was normal in patient III-3. Glycine 1389 is part of the Gly-Xaa-Yaa repeat, which is essential for the proper folding of the triple helix,18, 25 and is highly conserved through species and among all the six collagen IV genes. In silico analysis by Polyphen-2 and SIFT prediction programs indicated this change as probably damaging.
Family B: Subjects II-2, II-3, II-5, II-6, I-1 and I-2 were tested. In probands II-2, II-5 and subject I-2 COL4A2 sequencing showed a heterozygous c.3206delC deletion in exon 34 leading to a frameshift and premature stop at position +27 (Figure 1d). The COL4A2 sequence was normal in subjects I-1, II-3 and II-6. No DNA was available from patient II-1 and II-4. RT-PCR of the COL4A2 transcript in patient II-2's fibroblasts was performed. Only a transcript of the expected wild-type sequence was observed, compatible with degradation of the mutant mRNA by nonsense-mediated decay. Quantitative RT-PCR (RT-qPCR) of COL4A2 and COL4A1 transcript in fibroblasts from the same patient was performed and confirmed diminished amount of COL4A2 compared with COL4A1 mRNA (Figure 1e).
Skin transmission electron microscopy
Electron microscopy of the skin of patient II-2 from family B showed abnormalities of the epidermis–dermis junction with scattered thickening, blurring and duplications of the basement membrane (Figures 3b and c). These abnormalities were not observed in control skin (Figure 3a) and are similar to those seen in COL4A1 patients.8

Skin transmission electron microscopy. Upper arm skin biopsy of subject II-2 of family B was analysed by transmission electron microscopy according to Plaisier et al8 in double blind experiments by two investigators and independently scored. (a) Control skin biopsy showing illustrative digitations (arrows) of the epidermis–dermis junction with normal basement membrane structure. (b, c) Illustrative areas of the basement membrane of the epidermis-dermis junction in the patient shows areas of thickening, blurring, fragmentation and duplication, giving it at times a blebby appearance (arrows).
ER stress response and apoptosis
The mouse Col4a1+/Δ ex40 mutation leads to synthesis of abnormal Col4a1 chains, which accumulate in the ER of lens epithelia. This accumulation activates the unfolded protein response, a well-known ER stress response preceding apoptosis.20, 24, 26 As the c.3206delC COL4A2 mutation predicts a null-allele and no such mutations have been described in human COL4A1 patients, we investigated the pathogenic mechanism of this mutation in cultured skin fibroblasts from proband II-2. We wondered whether ER stress could be induced by abnormal stoichiometry of COL4A1 and COL4A2 chains, because of reduced COL4A2 synthesis.
ER stress response was analysed by staining fibroblasts with antibodies anti HSP47, a collagen-specific chaperon protein, and anti KDEL, an ER stress marker specific for ER-retained proteins. If collagen chains accumulate in patient cells, then increased storage of HSP47 can be expected.24, 26 Under standard culture conditions, HSP47 staining was comparable in patient and control cells, suggesting no accumulation of unfolded collagen chains (Supplementary Figure S3). Subsequently, we analysed whether KDEL staining under standard and stress conditions, induced by DTT, would detect ER stress in patient cells. Under the DTT condition, we know that normal cells slightly increase susceptibility to apoptosis from 10 to 18% (Figure 4c and Supplementary Figure S5).27 No clear signs of ER stress were detected by KDEL staining under DTT in the patient cells (Supplementary Figure S4).24 We then tested whether signs of expression of ER stress markers could be detected by western blot analysis using antibodies specific for IRE1, cleaved form of ATF6, CHOP, BiP and by real time-PCR for XBP-1 splicing.23 We did not detect expression of the markers in the patient cells, similarly to a control cell line (Figures 4a and b). As positive control, tunicamycin-treated control cells showed increased expression of markers, which means that the stress response was negative rather than below detection level. As ER stress is supposed to trigger apoptotic cell death, we studied sensitivity of the cells to apoptosis under standard conditions and stress stimuli. Under stress induced by DTT, we observed an abnormally high percentage (>60%) of apoptotic cells in the patient compared with five control cell lines (Figure 4c). No difference was observed in the absence of DTT treatment (Supplementary Figure S5). We conclude that COL4A23206delC fibroblasts do not show signs of ER-stress, although the patient-derived cells show more signs of stress-induced apoptosis.

COL4A2 c.3206delC mutation does not cause endoplasmic reticulum stress but reveals susceptibility to apoptosis. (a) Western blot analysis for endoplasmic reticulum stress markers (IRE-1, cleaved form of ATF6, CHOP and BiP) was negative in patient II-2 (family B) and control skin fibroblasts. (b) XBP-1 mRNA, which is spliced in response to endoplasmic reticulum stress, was evaluated by RT-PCR. Patient and control samples did not show detectable levels of spliced XBP-1. Tunicamycin treated cells were included as a positive control (+) in A and B. Untreated cells were used as a negative control (−). (c) Mean percentage of apoptotic cultured fibroblasts after stress induction by dithiothreitol. WRS: fibroblasts from patient with mutation in EIF2AK3 gene with increased susceptibility to apoptosis.27 COL4A2: cells from patient with c.3206delC mutation. Controls: fibroblast cell lines from five healthy individuals. Data are average of triplicate experiments.
Discussion
Pathogenesis of COL4A2 mutations
Using the knowledge of the Col4a2 mouse model, we have used a candidate gene approach in our porencephaly families, and we provide evidence that COL4A2 mutations represent a novel genetic risk factor for haemorrhagic porencephaly and small-vessel disease.
We describe cerebral abnormalities, including porencephaly, small-vessel disease in the form of scattered white matter lesions, carotid aneurysm, cerebellar and eye abnormalities in two families, associated with two dominantly inherited changes in COL4A2 (G1389R, c.3206delC), all in the triple helix domain of the procollagen IV alpha-2 protein. Carotid aneurysm and cerebellar hypoplasia have been observed in a single mutation carrier, respectively, in family A and B (Table 1), making a causal relationship possible but not definitive.
Extensive data from several types of collagens have shown that missense and splice site mutations in the triple helix domain are highly pathogenic and particularly substitutions of the glycine at each third position (G-Xaa-Yaa) disrupt the triple helix structure, with a dominant negative effect. These missense mutations lead to abnormal assembly of the triple helix with intracellular accumulation of procollagen chains.24 The stereotypical nature of pathogenic glycine missense mutations in collagens makes therefore mutation recognition relatively easy compared with other genes. Considering the cosegregation of the G1389R mutation with the cerebrovascular disease we conclude that this mutation is involved in the pathogenesis of the porencephaly and white matter lesions observed in family A.
The c.3206delC frame-shift mutation found in family B leads to loss of COL4A2 chain synthesis from one allele, however, the pathogenic mechanism might be different from the G-Xaa-Yaa type of mutation, as it is predicted to cause a loss of function and null-alleles have not been described in patients harbouring COL4A1-mutations.
In support of a pathogenic effect of the c.3206delC mutation, we observed an increased susceptibility to apoptosis and, in addition, EM of patient skin showed abnormalities of the epidermal basement membranes, suggesting abnormal extracellular collagen-IV assembly. Heterozygote truncating mutations have already been observed in collagen IV-related Alport syndrome, but null alleles for the C. elegans COL4A1 homologue, emb-9 and COL4A2 homologue let2 cause disruption of the basement membrane and embryonic lethality, although at a later stage than the G-Xaa-Yaa missense mutations.28
The non-collagen domain of collagen IV alpha-2, but not alpha-1 chain, promotes cell adhesion, outgrowth of embryonic neurons and is responsible for strict regulation of the 2:1 stoichiometry of the triple helix.18, 29, 30, 31
This could indicate that a disturbed ratio of procollagen alpha-1 and procollagen alpha-2 chains due to a heterozygous null allele of either COL4A1 or COL4A2 may lead to a disturbed or insufficient heterotrimer assembly and secretion.
The mechanism leading to stroke and porencephaly is presumably a decreased resistance of the vascular wall to increased mechanic stimuli. In the lens epithelia of Col4a1+/Δ ex40 mice accumulation of COL4A1 and COL4A2 in the ER induces an ER stress response, a known trigger of apoptosis.24 Nevertheless, studies supporting this mechanism in human COL4A1 mutations are lacking. We observed increased susceptibility of c.3206delC patient fibroblasts to apoptosis, possibly related to ER stress, which, however, could not be detected under our experimental conditions. Future studies are needed to address this discrepancy, which might be caused for example by the culture conditions compared with experiments performed in mouse tissues.20, 26 Apoptosis is an important mechanism regulating brain development and is involved in the onset of microcephaly.32 Some of the COL4A2 patients are born microcephalic and the reason is not apparent at the moment, but it could be related to susceptibility of the developing brain to apoptosis.27 It is also possible that, independently of the ER stress, COL4A2 and the apoptotic pathways are linked as they are developmentally co-regulated. In fact, it has been shown that micro RNA mir-29b represses expression of both COL4A2 and antiapoptotic Bcl-2 family member Mcl-1 during skeletal development.33, 34
COL4A2 mutation: phenotypic variation and reduced penetrance
COL4A1 and COL4A2 are ubiquitously co-expressed in the basement membrane of epithelia and endothelia and from the observation of mouse Col4a1 and Col4a2 mutations, human COL4A1 and COL4A2 mutations could be predicted to result in cerebral bleeding. However, only COL4A1 mutations have been identified after linkage in large pedigrees.3 One of the reasons could be the difficulty to recruit large families with similarly affected patients, because of phenotypic variability or reduced penetrance of COL4A2 mutations, similarly to other causes of stroke and aneurysm,35 which makes linkage analysis quite difficult.
We can assume reduced penetrance of the COL4A2 mutations in both our families.
In family A subject II-2 has the G1389R mutation and is asymptomatic, but MRI revealed a carotid aneurysm, similar to the findings in an obligate carrier of COL4A1 mutation that we previously described.7
In family B, we also observed an asymptomatic carrier (subject I-2) of the c.3206delC COL4A2 mutation, who presents ocular anomalies but denied an MRI. Asymptomatic carriers have been observed also in COL4A1 mutation and, although this has been interpreted as reduced penetrance of the mutation, extensive investigation has often revealed hidden signs of the disease.3, 7 However, considering the wide heterogeneity of symptoms, the contribution of other factors, besides the COL4A1 mutation, has been suggested as trigger of the pathogenic event.3
In both our families, a wide intra-familial phenotypic variation has been observed, similar to COL4A1 mutations.7 In Col4a1 mutant mice it has been shown that the genetic background is important to determine the phenotype, probably through the effect of modifying loci.24 Col4a2 mutant mice present with cerebral porencephaly, microphthalmia, lens opacity, buphthalmos and renal anomalies, but in general with a milder phenotype compared with Col4a1-deficient mice.19
Interestingly, in family B we observed an individual (subject II-6), affected by a perinatal intraparenchymal bleeding leading to a cyst, who lacks the familial COL4A2 mutation (Supplementary Figure S2) and additional children died at young age with severe cerebral haemorrhage and hydrocephalus, who were not tested for COL4A2 mutations. Subject II-6 is heterozygote for protein S deficiency, which in itself has not been the cause of the bleeding but might have contributed to the onset of the germinal matrix haemorrhage. As for COL4A1 mutation carriers, it is therefore possible that in this family additional factors have summed up and contribute to the phenotypic manifestation.
The particular fragility of the cerebral vessels in patients with either COL4A1 or COL4A2 mutations remains unexplained in light of the ubiquitously expression of COL4A1 and COL4A2. However, it could reflect selective expression in subtypes of brain vascular endothelia.4 It has been shown that in the kidney expression of COL4A3, COL4A4 and COL4A5 genes can be cell-type specific.36, 37
In summary, we observed that familial COL4A2 mutations could lead to a wide spectrum of cerebrovascular disorders, including porencephaly, white matter lesions and possibly aneurysm and cerebellar hypoplasia, which might go undiagnosed because of reduced penetrance and variable expression. Additional risk factors may influence the course of cerebrovascular diseases in families with COL4A2 mutations.
References
Dichgans M : Genetics of ischaemic stroke. Lancet Neurol 2007; 6: 149–161.
Domingues-Montanari S, Mendioroz M, del Rio-Espinola A, Fernandez-Cadenas I, Montaner J : Genetics of stroke: a review of recent advances. Expert Rev Mol Diagn 2008; 8: 495–513.
Gould DB, Phalan FC, Breedveld GJ et al: Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 2005; 308: 1167–1171.
Govaert P : Prenatal stroke. Semin Fetal Neonatal Med 2009; 14: 250–266.
Tonk M, Haan J : A review of genetic causes of ischemic and hemorrhagic stroke. J Neurol Sci 2007; 257: 273–279.
Vahedi K, Alamowitch S : Clinical spectrum of type IV collagen (COL4A1) mutations: a novel genetic multisystem disease. Curr Opin Neurol 2011; 24: 63–68.
Breedveld G, de Coo IF, Lequin MH et al: Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly. J Med Genet 2006; 43: 490–495.
Plaisier E, Gribouval O, Alamowitch S et al: COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med 2007; 357: 2687–2695.
van der Knaap MS, Smit LM, Barkhof F et al: Neonatal porencephaly and adult stroke related to mutations in collagen IV A1. Ann Neurol 2006; 59: 504–511.
Volonghi I, Pezzini A, Del Zotto E et al: Role of COL4A1 in basement-membrane integrity and cerebral small-vessel disease. The COL4A1 stroke syndrome. Curr Med Chem 2010; 17: 1317–1324.
Meuwissen ME, de Vries LS, Verbeek HA et al: Sporadic COL4A1 mutations with extensive prenatal porencephaly resembling hydranencephaly. Neurology 2011; 76: 844–846.
Coupry I, Sibon I, Mortemousque B, Rouanet F, Mine M, Goizet C : Ophthalmological features associated with COL4A1 mutations. Arch Ophthalmol 2010; 128: 483–489.
Gould DB, Phalan FC, van Mil SE et al: Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med 2006; 354: 1489–1496.
Sibon I, Coupry I, Menegon P et al: COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann Neurol 2007; 62: 177–184.
Vahedi K, Boukobza M, Massin P, Gould DB, Tournier-Lasserve E, Bousser MG : Clinical and brain MRI follow-up study of a family with COL4A1 mutation. Neurology 2007; 69: 1564–1568.
Plaisier E, Chen Z, Gekeler F et al: Novel COL4A1 mutations associated with HANAC syndrome: a role for the triple helical CB3[IV] domain. Am J Med Genet A 2010; 152A: 2550–2555.
Vahedi K, Kubis N, Boukobza M et al: COL4A1 mutation in a patient with sporadic, recurrent intracerebral hemorrhage. Stroke 2007; 38: 1461–1464.
Khoshnoodi J, Pedchenko V, Hudson BG : Mammalian collagen IV. Microsc Res Tech 2008; 71: 357–370.
Favor J, Gloeckner CJ, Janik D et al: Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics 2007; 175: 725–736.
Marutani T, Yamamoto A, Nagai N, Kubota H, Nagata K : Accumulation of type IV collagen in dilated ER leads to apoptosis in Hsp47-knockout mouse embryos via induction of CHOP. JCell Sci 2004; 117: 5913–5922.
Verkerk AJ, Schot R, Dumee B et al: Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet 2009; 85: 40–52.
Lobel U, Sedlacik J, Gullmar D, Kaiser WA, Reichenbach JR, Mentzel HJ : Diffusion tensor imaging: the normal evolution of ADC, RA, FA, and eigenvalues studied in multiple anatomical regions of the brain. Neuroradiology 2009; 51: 253–263.
Lin JH, Li H, Yasumura D et al: IRE1 signaling affects cell fate during the unfolded protein response. Science 2007; 318: 944–949.
Gould DB, Marchant JK, Savinova OV, Smith RS, John SW : Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum Mol Genet 2007; 16: 798–807.
Engel J, Prockop DJ : The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper. Annu Rev Biophys Biophys Chem 1991; 20: 137–152.
Firtina Z, Danysh BP, Bai X, Gould DB, Kobayashi T, Duncan MK : Abnormal expression of collagen IV in lens activates unfolded protein response resulting in cataract. J Biol Chem 2009; 284: 35872–35884.
Poulton CJ, Schot R, Kia SK et al: Microcephaly with simplified gyration, epilepsy, and infantile diabetes linked to inappropriate apoptosis of neural progenitors. Am J Hum Genet 2011; 89: 265–276.
Gupta MC, Graham PL, Kramer JM : Characterization of alpha1(IV) collagen mutations in Caenorhabditis elegans and the effects of alpha1 and alpha2(IV) mutations on type IV collagen distribution. J Cell Biol 1997; 137: 1185–1196.
Herbst TJ, McCarthy JB, Tsilibary EC, Furcht LT : Differential effects of laminin, intact type IV collagen, and specific domains of type IV collagen on endothelial cell adhesion and migration. J Cell Biol 1988; 106: 1365–1373.
Petitclerc E, Boutaud A, Prestayko A et al: New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem 2000; 275: 8051–8061.
Lein PJ, Higgins D, Turner DC, Flier LA, Terranova VP : The NC1 domain of type IV collagen promotes axonal growth in sympathetic neurons through interaction with the alpha 1 beta 1 integrin. J Cell Biol 1991; 113: 417–428.
Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB : A developmental and genetic classification for malformations of cortical development. Neurology 2005; 65: 1873–1887.
Li Z, Hassan MQ, Jafferji M et al: Biological functions of miR-29b contribute to positive regulation of osteoblast differentiation. J Biol Chem 2009; 284: 15676–15684.
Mott JL, Kobayashi S, Bronk SF, Gores GJ : mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007; 26: 6133–6140.
Guo DC, Papke CL, Tran-Fadulu V et al: Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet 2009; 84: 617–627.
Abrahamson DR, Hudson BG, Stroganova L, Borza DB, St John PL : Cellular origins of type IV collagen networks in developing glomeruli. J Am Soc Nephrol 2009; 20: 1471–1479.
Heidet L, Cai Y, Guicharnaud L, Antignac C, Gubler MC : Glomerular expression of type IV collagen chains in normal and X-linked Alport syndrome kidneys. Am J Pathol 2000; 156: 1901–1910.
Acknowledgements
We thank Dr Hajo Wildschut for prenatal patient care, and Dr Marion Smit, Ton de Jong, Ronald Grefhorst, Tom de Vries-Lentsch and Ruud Koppenol for their logistic and technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Verbeek, E., Meuwissen, M., Verheijen, F. et al. COL4A2 mutation associated with familial porencephaly and small-vessel disease. Eur J Hum Genet 20, 844–851 (2012). https://doi.org/10.1038/ejhg.2012.20
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.20
Keywords
This article is cited by
-
Cold-induced vasodilation response in a Japanese cohort: insights from cold-water immersion and genome-wide association studies
Journal of Physiological Anthropology (2023)
-
A Novel Mutation in COL4A1 Gene in a Chinese Family with Pontine Autosomal Dominant Microangiopathy and Leukoencephalopathy
Translational Stroke Research (2022)
-
Novel heterozygous COL4A2 variant c.2572A > G, p.(I858V) mimicking Sneddon’s and Divry van Bogaert Syndrome
Journal of Neurology (2022)
-
Deletion in COL4A2 is associated with a three-generation variable phenotype: from fetal to adult manifestations
European Journal of Human Genetics (2021)
-
MFAP2 is overexpressed in gastric cancer and promotes motility via the MFAP2/integrin α5β1/FAK/ERK pathway
Oncogenesis (2020)
