
Abstract
Inherited retinal degeneration (IRD) is a common cause of visual impairment (prevalence ∼1/3500). There is considerable phenotype and genotype heterogeneity, making a specific diagnosis very difficult without molecular testing. We investigated targeted capture combined with next-generation sequencing using Nimblegen 12plex arrays and the Roche 454 sequencing platform to explore its potential for clinical diagnostics in two common types of IRD, retinitis pigmentosa and cone-rod dystrophy. 50 patients (36 unknowns and 14 positive controls) were screened, and pathogenic mutations were identified in 25% of patients in the unknown, with 53% in the early-onset cases. All patients with new mutations detected had an age of onset <21 years and 44% had a family history. Thirty-one percent of mutations detected were novel. A de novo mutation in rhodopsin was identified in one early-onset case without a family history. Bioinformatic pipelines were developed to identify likely pathogenic mutations and stringent criteria were used for assignment of pathogenicity. Analysis of sequencing metrics revealed significant variability in capture efficiency and depth of coverage. We conclude that targeted capture and next-generation sequencing are likely to be very useful in a diagnostic setting, but patients with earlier onset of disease are more likely to benefit from using this strategy. The mutation-detection rate suggests that many patients are likely to have mutations in novel genes.
Similar content being viewed by others


Introduction
Inherited retinal degeneration (IRD) is a term used to describe a wide range of genetically determined retinal disorders, which cause varying degrees of progressive visual dysfunction and blindness. The nosology of IRD is complex, ranging from eponymous classifications to clinical and functional phenotyping using imaging, electrophysiology and psychophysical evaluation.
Many recognisable phenotypes are associated with mutations in specific genes, for example ABCA4 in Stargardt disease, but retinitis pigmentosa and rod-cone/cone-rod dystrophies exhibit significant genetic heterogeneity ( https://sph.uth.tmc.edu/retnet – last accessed 23 March 2012). These cases provide the greatest challenge for clinicians in providing accurate prognosis and genetic recurrence risks to family members. The frequency of mutations in each of these genes is unknown, and in a diagnostic setting sequential genetic testing might be considered. Hitherto, genetic testing technologies have been limited to Sanger sequencing, which is very accurate but prohibitively costly for large-scale sequencing, or screening methods such as APEX arrays which detect only known mutations. Because of these complex clinical, technical and costing issues, very few genes have been available for testing within a clinical diagnostic setting in the UK or elsewhere (Supplementary Table 1).1, 2 It is recognised that genetic testing has an increasingly important role in IRD, not only for clinical diagnostics and for prognosis but also for potential therapeutic interventions such as gene therapy, and since High Throughput or next-generation sequencing (NGS) allows multiple genes to be sequenced in parallel, this could be used to increase molecular diagnostic rates in IRD. As IRD is highly clinically and genetically heterogeneous, it offers an ideal model in which to investigate the complexities of introducing NGS into a clinical diagnostic setting. We therefore set out to identify pathogenic mutations in a cohort of patients with retinitis pigmentosa or cone-rod dystrophy from an Ophthalmic Genetic clinic to determine which patients would be most likely to benefit from a targeted screening approach in a clinical diagnostic setting. There have been two other recent publications using NGS in retinal dystrophies. The first reported a detection rate of 57% in 20 patients with a mixture of retinal phenotypes and the second reported a 36% detection rate in 100 patients with retinitis pigmentosa. However, neither reported data on the age of onset of the condition in the patients who were analysed.3, 4 We used targeted capture combined with NGS to enrich and sequence for genes of interest, to determine whether NGS could be a useful introduction into clinical practice and, if so, which patient groups would be most likely to benefit.
Patients and methods
Human subjects
All patients (and involved relatives) were counselled and consented according to the Declaration of Helsinki and the study was approved by the local Ethics Committee.5 Patients were recruited if they were diagnosed with retinitis pigmentosa (RP) or cone-rod dystrophy (CRD) phenotype in a specialist Ophthalmic Genetics Clinic (run by SMD). We excluded any patient from this analysis who did not have RP or CRD, or was considered to have a multifactorial condition such as age-related macular degeneration. We also excluded patients with likely Stargardt disease, who were analysed in a separate study.6
Fifty-five patients were targeted and sequenced, 50 of which were included in the final analysis (5 were excluded because of low coverage – see results). 14/50 samples were positive controls. The positive controls included 12 patients who had been screened for ABCA4 mutations in a previous study and 2 patients with heterozygous dominant mutations identified in PRPF8 and PRPF31.6 Nineteen pathogenic variants were known in the positive control group (Supplementary Table 2).
Targeted capture and next-generation sequencing
A custom NimbleGen 12-plex capture array was designed and manufactured by Roche NimbleGen. The array was designed to capture 1413 exons from 73 IRD genes and 1 deep intronic splice site in CEP290, known to be associated with LCA (c.2991+1655A>G) (Supplementary Table 3).7 The design also included the entire genomic region of ABCA4 as some patients with a single mutation in ABCA4 might harbour mutations in the intronic region, although only the 25 bp of intronic sequence surrounding the exons was included in the current analysis. The target regions of interest (ROI), including exons and UTRs were identified and compiled using NCBI build 36.1, hg18 and the Ensembl database, and all annotated transcripts were included. The final NimbleGen design was 463.4 kb in size and was made up of 135 000 (135K) overlapping 60–90 bp probes that covered 96.4% ROI. Genes were included if they were known to have previously been associated with IRD in the literature or RetNet (https://sph.uth.tmc.edu/retnet – last accessed 23 March 2012).
Library preparations were made using the Roche manufacturer’s protocol. In brief, 7 μg of patient’s genomic DNA was sheared by nebulisation into fragments of 400–800 bp (mean 600 bp). The fragment ends were polished using T4 PNK and T4 DNA polymerase and then adaptors containing a unique 454 Multiple Identifier Sequence (MID) were ligated. The library was immobilised to streptavidin beads to allow elution of single-stranded DNA, which was amplified by linker-mediated PCR using adaptor-specific primers.
An amplified library of 1 μg was loaded onto 1 region of the 12-plex array with 100 μg of COT-1 and MID-specific hybridisation oligonucleotides. Each region of the array contained the full 135K probe design. The array was hybridised for 60–70 h, washed and eluted as per NimbleGen instructions. The captured DNA was then re-amplified using linker-mediated PCR.
Emulsion PCR was performed using an optimal ratio of DNA to capture beads as per manufacturer’s instructions. The emulsion was broken, the capture beads containing DNA were recovered and the sequencing primers were added. These samples were sequenced on 2 × 454 Titanium runs: each run used the two-lane gasket option and the same 12-plex capture was run on both lanes.
Sequence-read mapping and variant calling
Read data were analysed using the 454 Sequencing System Software package (version 2.5.3). Sequencing run files (SFF format) were split by multiplex identifiers (MID) using the ‘sfffile’ program. The GS Reference Mapper software with default parameters was then used to map sequence reads to the human reference genome (NCBI build 36, hg18) and to detect variants. Identified variants were annotated through the refGene database and flagged if already present in dbSNP (build 130). Only high-confidence variants (ie, those reported in the ‘454HCDiff.txt’ output file) within the targeted regions were considered.
Sanger sequencing validation
All mutations identified by NGS and considered likely to be pathogenic were validated by Sanger sequencing using standard protocols and where possible family members were tested to determine phase and investigate cosegregation of the variants with disease.
Sequence coverage was estimated from the ‘454ReadStatus.txt’ output file with custom perl scripts. Only reads with mapping status ‘Full’ or ‘Partial’ were included in the analysis. The following metrics were determined: total data per array, percentage of total reads aligned to the ROI and for each ROI: minimum, maximum and mean number of reads and percentage of bases with ≥10 × , 20 × , 30 × coverage. These data were used to determine whether there was any bias of coverage in samples with or without mutations identified.
Variants were annotated using the analysis pipeline detailed in Figure 1. This pipeline included pathogenicity prediction programs PolyPhen2, SIFT and MutPred in addition to splice prediction programs MaxEntScan, NNSplice, Human Splice site finder and Splice Site finder.8, 9, 10, 11, 12, 13 Missense mutations were considered likely to be pathogenic if (a) PolyPhen2, SIFT and MutPred all suggested pathogenicity (b) the variant has been previously reported with good evidence for a functional effect (c) low frequency in the general population and (d) in recessive cases the second mutation was found. We took a very conservative approach to considering missense mutations as pathogenic, because we have data from the analysis of rhodopsin to suggest that previously reported pathogenic mutations are in fact benign polymorphisms.14 Patient data were analysed by age of onset, presence or absence of family history and the phenotype. We calculated the percentage of bp of submitted exons that were missing from the NimbleGen final probe design. We also identified the number and percentage of exons missing from the final sequence data. We also analysed four samples with equivalent total reads and alignment to investigate the contribution of capture efficiency to the variability in overall depth of coverage obtained.

Data-analysis pipeline; the approximate number of variants at each analysis stage is shown.
Results
Samples of 55 patients were captured on a total of 8 arrays and sequenced in a total of 17 runs. In 5 of the 55 samples <80% of bases were covered at 10 × and the samples were excluded from further analysis. Data metrics for the remaining 50 samples (36 unknown IRD, 14 controls) are shown in Table 1.
Identification of mutations in positive controls
Eighteen of the 19 pathogenic mutations present in the 14 control samples were detected (Supplementary Table 2). The mutation that was not detected, ABCA4 c.4537dupC in exon 30, is within a short homopolymer tract of 7 C (Supplementary Figure 1). The coverage of this base in this sample was 38 × .15
Identification of variants considered likely to be pathogenic in patients with an unknown retinal degeneration
Mutations that were considered highly likely to be pathogenic, based on our analysis pipeline, were found in 9/36 patients in a total of 6 genes, making a total pathogenic mutation-detection rate of 25% (Table 2). In the patients with an age of onset under 21 the detection rate was 53% (Supplementary Table 5). The mutations were located at 13 different positions, of which 4 were novel. In total, 6 of the 9 patients had two autosomal recessive mutations, and 3 of the 9 patients had 1 autosomal dominant mutation. Surprisingly, 1 early-onset patient had the mutation P53R in Rhodopsin, which has only been described once before, segregating in an autosomal dominant RP family with adult-onset.16 This mutation was shown to be de novo, when parental samples were analysed (Table 2 and Supplementary Table 4) and analysis of photopigment function and cellular trafficking confirmed the pathogenicity of this variant.14 There is one very recent report of a de novo mutation in rhodopsin, although in that report, the data were inconclusive because paternal DNA was unavailable.4 Of the presumed pathogenic variants identified, 31% were present in dbSNP, demonstrating that this database likely contains numerous pathogenic variants. A further patient had a single mutation: 2465fsX2 in exon 54 of CEP290 (which causes autosomal recessive disease), which was considered highly likely to be pathogenic, but the second mutation was not found. Overall, in the mutation-positive patients only 5 of the 13 mutations were missense mutations, reflecting the very conservative assessment we made of pathogenicity.
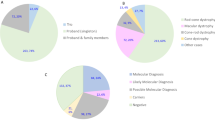
All patients with identifiable mutations had an age of onset <21 years and 44% had a positive family history: 2 with a dominant family history, and 2 with a family history suggestive of recessive inheritance (affected siblings or consanguinity). In the patients without identifiable mutations, 70% had an age of onset over 21 years and 78% did not have any relevant family history (Figure 2 and Supplementary Table 5).

Age range and family history status of patients. (a) Age range of patients with pathogenic mutation(s) identified. (b) Age range of patients without pathogenic mutation(s) identified. (c) Family history in patients with pathogenic mutations identified (d) Family history in patients without a pathogenic mutation identified (e) Distribution of phenotypes in patients with a pathogenic mutation (red) compared with the total (blue).
In the patient with the CEP290 deletion in exon 54 that produced a premature truncation codon (Table 2), no second mutation could be found, even in the cryptic splice site. The coverage for CEP290 in this patient was over 99% of bases covered by at least 10 × and 82% covered by at least 30 × , suggesting that low coverage is not the explanation for the missing second mutation.
When the records of all the cases were reviewed retrospectively, the mutations were considered to be consistent with the phenotype. A total of 19 variants were checked by Sanger sequencing of which three were not verified. Two had a low ratio of variant to reference sequence, and the third was an insertion, suggesting that all were artefacts.
Capture and sequence coverage variability
In all, 4.6% of exons, including 13 full exons, were not covered by the NimbleGen probes. However, we found that not all the regions were consistently missing from the sequence data, presumably because some of the regions are captured by neighbouring probes. One exon in PITPNM3 was consistently and entirely missed from the final data despite being present in the design. We compared 4 samples with similar levels of coverage (61–71 × ) and found that there was considerable variation in the base pairs and exons that were missing (Table 3). For example: 93–97% of base pairs were covered at 10 × and 94–98% of exons were completely covered (10 × or more). However, if coverage at 30 × is considered as the gold standard, then only 71–82% of exons were covered at this depth. Although there was a general correlation between missing exons and depth of coverage, this was not linear: our conclusions from this analysis are that for a specific depth of coverage, there is significant variability in the ROI with missing data.
Analysis comparing different patient-group metrics
When the data were analysed for percentage coverage at <10, <20 and <30 × , we found that there was no evidence of bias towards the identification of pathogenic mutations in regions of greater coverage (Figure 3). The positive control samples showed a similar spread of coverage compared with the probands, suggesting that there is no specific bias in our data set to account for some patients not having mutations detected (Figure 3).

Sample coverage. The percentage of bases covered at 10 × , 20 × and 30 × for each patient. Patients with mutation(s) identified (blue), patients with no identified mutations (black), control patients (green) and patients with <than 80% of bases covered at 10 × (yellow).
Discussion
We used targeted NGS in patients with retinitis pigmentosa and cone-rod dystrophy to address the issues specifically related to clinical diagnostics, namely: (a) the characteristics of patients most likely to have mutations detected in order to provide guidelines for genetic testing, (b) genotype–phenotype correlations, (c) determining important sequencing metrics, and (d) bioinformatic challenges of determining pathogenicity in the context of large number of sequence variants. These issues are particularly relevant as targeted sequencing is likely to be the technology of choice for diagnostic purposes. This is because whole exome/genome-analysis pipelines need to be refined further to provide the accuracy required in a diagnostic setting.
We found that certain patient categories increased the likelihood of detecting mutations in our gene set. The most important was the age of onset: all the patients with mutations had an onset under 21 years of age. This skewing of mutation detection towards the earlier age of onset group in our patients suggests that routine screening of patients over the age of 50 in a diagnostic setting is probably not warranted until further data are available. The explanation for skewing towards cases with an onset of under 21 years of age is unclear, but it may be that cases with an onset of over 50 years of age have a multigenic or multifactorial aetiology including accumulated environmental factors.
A positive family history was a poorer predictor in identifying a mutation. Although simplex cases are often considered most likely to represent autosomal recessive disease, we found a de novo dominant mutation in one patient, illustrating that it is important to include this possibility when providing genetic counselling. 8/17 (47%, Supplementary Table 5) cases with an onset of under 21 did not have mutations detected in contrast to the 53% of who did. Further work is in progress to investigate this, using whole-exome sequencing to determine if these patients have mutations in novel genes.
Although the phenotype was retrospectively considered to be consistent with the mutations found, this could not have been predicted a priori, illustrating the benefits of sequencing multiple genes in parallel. This was particularly the case for ABCA4 mutations in which the patients had atypical non-STGD phenotypes, a well-recognised phenomenon with ABCA4 retinopathies.17 We found 1 patient with a single-CEP290 mutation. Mutations in this exon are known to cause Joubert and Meckel syndromes and LCA, so we considered this mutation highly likely to be pathogenic, but we were unable to identify the 2nd mutation despite good coverage across the gene.7, 26 Thirty-one percent of the mutations we identified were novel, indicating that screening methods, which only detect for known mutations, would have missed a significant number of these in our data set.
We examined metrics that will be relevant when introducing NGS into clinical diagnostics. In particular, we examined metrics that might affect accuracy of the result, given that in a diagnostic setting this is of crucial importance. We found that there was variability in the capture efficiency, reflected by the variability of alignment (10–76%, average 58%), which is similar to other reports of hybridisation-based capture.27 We also found that average depth of coverage was highly variable (24–117 × , average 58 × ). Even when we controlled for the coverage, we found that the ROI that were missing were highly variable and not consistent, with only one exon that was entirely missed in all samples. A specific problem we noted was the difficulty of detecting mutations in homopolymer tracts using the 454 pyrosequencing chemistry. We show here that this is a problem with the 454 software rather than the chemistry itself, as the mutation was detected, but failed on quality scores. However, altering the software to increase mutation detection will increase the number of false positives leading to the need for additional validation by Sanger sequencing.
A major consideration for diagnostics is determining pathogenicity in the context of large number of sequence variants. Our analysis pipeline required several modifications during the analysis process. We found that if we excluded variants present in dbSNP during the filtering process, we removed 31% of the likely pathogenic variants in our data set. This applied to both recessive and dominant mutations. We also found that when synonymous variants were excluded, a splice mutation was missed. Therefore we adjusted our filtering algorithms to include splice predictions for synonymous variants and to include variants in dbSNP. We also noted that some indels were miscalled by the 454 software as synonymous SNPs and these were only identified manually. Use of commercially available software programmes may resolve some of these issues.
To assess pathogenicity of individual variants, we used standard pathogenicity prediction programmes, and missense variants that were considered to be benign by any of the programs were removed. This strategy reduced the number of variants to be analysed, but may have resulted in some pathogenic variants being removed and therefore missed during the filtering process. We were particularly conservative in our assessment of missense mutations, as we have recently found missense mutations in rhodopsin that are classified and reported as pathogenic by prediction programs but are almost certainly benign polymorphisms.14 However, this is not a new problem, as it is well recognised that bioinformatics prediction programs have limitations.
Two other papers have published data on the use of NGS for diagnostics in retinal degeneration with detection rates varying from 25% in our current study to 57%. These differences in detection rates are almost certainly due to variations in patient selection criteria, although it is possible that our lower detection rate is also due to our very conservative approach in making estimates of pathogenicity. Specifically, Neveling et al,8 considered missense variants to be pathogenic, if two of the three programs predicted pathogenicity, whereas we required all three programs to consider a missense variant to be pathogenic. These differences emphasise that patient-selection criteria are of utmost importance in maximising the detection rate in a diagnostic setting and also that interpretation of pathogenicity remains a challenge for diagnostic services.14
Conclusions
We have shown that NGS offers significant advantages over conventional Sanger sequencing or screening technologies for clinical diagnostics of RP/CRD. However, we also show that capture methods, sequencing chemistries and bioinformatics pipelines need to be optimised to use NGS in a clinical diagnostic setting with confidence. Until these are achieved, the following points should be considered:
-
Genetic testing guidelines and strategies to determine which patients are most likely to benefit from testing will become clearer as NGS use becomes more widespread. Our data suggest that targeting testing towards those with a younger age of onset or a family history may be the most cost effective. When reporting that ‘no mutation has been identified’, it should be made clear that some mutations may not be detected owing to factors such as lack of depth of coverage and miscalling of bases; as NGS evolves these problems will be addressed.
-
All variants should be validated by Sanger sequencing before they are formally reported in the clinical setting.
References
Jaakson K, Zernant J, Külm M et al: Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat 2003; 22: 395–403.
Klevering BJ, Yzer S, Rohrschneider K et al: Microarray-based mutation analysis of the ABCA4 (ABCR) gene in autosomal recessive cone-rod dystrophy and retinitis pigmentosa. Eur J Hum Genet 2004; 12: 1024–1032.
Audo I, Bujakowska K, Leveillard T et al: Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis 2012; 7: 8.
Neveling K, Collin RW, Gilissen C et al: Next generation genetic testing for retinitis pigmentosa. Hum Mutat 2012; 33: 963–967.
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. J Postgrad Med 2002; 48: 206–208.
Downes SM, Packham ER, Cranston T, Clouston P, Seller A, Nemeth AH : Detection rate of pathogenic mutations in ABCA4 using direct sequencing: clinical and research implications. Arch Ophthalmol, (in press).
den Hollander AI, Koenekoop RK, Yzer S et al: Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet 2006; 79: 556–561.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Ng PC, Henikoff S : Predicting deleterious amino acid substitutions. Genome Res 2001; 11: 863–874.
Li B, Krishnan V, Mort ME et al: Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 2009; 25: 2744–2750.
Yeo G, Burge CB : Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004; 11: 377–394.
Reese MG, Eeckman FH, Kulp D, Haussler D : Improved splice site detection in Genie. J Comput Biol 1997; 4: 311–323.
Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C : Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009; 37: e67.
Davies WIL, Downes SM, Fu JK et al: Next Generation Sequencing (NGS) in healthcare delivery: lessons from the functional analysis of rhodopsin. Genet Med 2012; e-pub ahead of print 12 July 2012; doi:10.1038/gim.2012.73.
Margulies M, Egholm M, Altman WE et al: Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005; 437: 376–380.
Inglehearn CF, Keen TJ, Bashir R et al: A completed screen for mutations of the rhodopsin gene in a panel of patients with autosomal dominant retinitis pigmentosa. Hum Mol Genet 1992; 1: 41–45.
Maugeri A, Klevering BJ, Rohrschneider K et al: Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet 2000; 67: 960–966.
Shroyer NF, Lewis RA, Lupski JR : Complex inheritance of ABCR mutations in Stargardt disease: linkage disequilibrium, complex alleles, and pseudodominance. Hum Genet 2000; 106: 244–248.
September AV, Vorseter AA, Ramesar RS, Greenberg LJ : Mutation spectrum and founder chromosomes for the ABCA4 gene in South African patients with Stargardt disease. Invest Ophthalmol Vis Sci 2004; 45: 1705–1711.
Zernant J, Schubert C, Im KM et al: Analysis of the ABCA4 gene by next-generation sequencing. Invest Ophthalmol Vis Sci 2011; 52: 8479–8487.
McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP : Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA 1995; 92: 3249–3253.
Weigell-Weber M, Fokstuen S, Török B, Niemeyer G, Schinzel A, Hergersberg M : Codons 837 and 838 in the retinal guanylate cyclase gene on chromosome 17p: hot spots for mutations in autosomal dominant cone-rod dystrophy? Arch Ophthalmol 2000; 118: 300.
McLaughlin ME, Sandberg MA, Berson EL, Dryja TP : Recessive mutations in the gene encoding the −subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet 1993; 4: 130–134.
den Hollander AI, ten Brink JB, de Kok YJ et al: Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat Genet 1999; 23: 217–221.
den Hollander AI, Davies J, van der Velde-Visser SD et al: CRB1 mutation spectrum in inherited retinal dystrophies. Hum Mutat 2004; 24: 355–369.
Brancati F, Barrano G, Silhavy JL et al: CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am J Hum Genet 81: 104–113.
Hoppman-Chaney N, Peterson LM, Klee EW, Middha S, Courteau LK, Ferber MJ : Evaluation of oligonucleotide sequence capture arrays and comparison of next-generation sequencing platforms for use in molecular diagnostics. Clin Chem 2010; 56: 1297–1306.
Acknowledgements
This work was supported by the Oxford Partnership Comprehensive Biomedical Research Centre with funding from the Department of Health’s NIHR Biomedical Research Centre Programme. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health. This work was also supported in part by Roche Diagnostics, AK was funded by Ataxia UK, and JB and JR were funded by The Wellcome Trust (grant 075491/Z/04). WILD was supported by a Biotechnology and Biological Sciences Research Council (BBSRC) grant to MWH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Shanks, M., Downes, S., Copley, R. et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur J Hum Genet 21, 274–280 (2013). https://doi.org/10.1038/ejhg.2012.172
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.172
Keywords
This article is cited by
-
Genetic characteristics of 234 Italian patients with macular and cone/cone-rod dystrophy
Scientific Reports (2022)
-
Targeted next generation sequencing and family survey enable correct genetic diagnosis in CRX associated macular dystrophy – a case report
BMC Ophthalmology (2021)
-
Genetic characteristics and epidemiology of inherited retinal degeneration in Taiwan
npj Genomic Medicine (2021)
-
Unmasking Retinitis Pigmentosa complex cases by a whole genome sequencing algorithm based on open-access tools: hidden recessive inheritance and potential oligogenic variants
Journal of Translational Medicine (2020)
-
The mutation spectrum in familial versus sporadic congenital cataract based on next-generation sequencing
BMC Ophthalmology (2020)
