
Abstract
Population isolates are a valuable resource for medical genetics because of their reduced genetic, phenotypic and environmental heterogeneity. Further, extended linkage disequilibrium (LD) allows accurate haplotyping and imputation. In this study, we use nuclear and mitochondrial DNA data to determine to what extent the geographically isolated population of the Val Borbera valley also presents features of genetic isolation. We performed a comparative analysis of population structure and estimated effective population size exploiting LD data. We also evaluated haplotype sharing through the analysis of segments of autozygosity. Our findings reveal that the valley has features characteristic of a genetic isolate, including reduced genetic heterogeneity and reduced effective population size. We show that this population has been subject to prolonged genetic drift and thus we expect many variants that are rare in the general population to reach significant frequency values in the valley, making this population suitable for the identification of rare variants underlying complex traits.
Similar content being viewed by others


Introduction
The use of population isolates has proven valuable to map loci coding for complex traits (eg, Holm et al,1 Sulem et al2 and Thorgeirsson et al3). Genetic isolates present key features that simplify gene mapping, namely reduced phenotypic and environmental variance, and reduced genetic heterogeneity.4, 5 Genomes of individuals from isolated populations tend to be more homogeneous compared with other populations, reflected by a small effective population size (Ne or the effective number of individuals required to explain the observed genetic variability).6 In population isolates, a small Ne may arise as a consequence of a founding event (ie, the settlement of a new territory) and it is maintained through time owing to the absence of gene flow (migration) with neighbouring populations. In this scenario genetic drift (the random fluctuation of allele frequency at each generation) can lead to quick significant reduction of extant variability and the frequency of disease or trait-associated variants can increase because of drift, thus facilitating gene mapping.7
Another key property of population isolates is the large extension of regions in linkage disequilibrium (LD).8 Isolates are relatively young compared with the population of origin, and usually originated from a small founding nucleus of individuals, two conditions that create association between loci that are far apart from each other. In addition, because of the small Ne often recombination takes place between identical haplotypes, further increasing the range of significant LD. As a consequence, any two individuals in the population tend to share potentially long chromosomal segments identical by descent, facilitating long-range haplotype matching, genotype imputation9, 10, 11 and reconstruction of population-specific recombination maps.
The Val Borbera is a geographically isolated valley within the Appennine Mountains of Piedmont (North-West Italy). According to genealogical records, about 3000 individuals (the majority of the current population) descend from inhabitants of the valley in the 17th century. Previous demographic and epidemiological analyses highlight features characteristic of genetic isolates, including a high percentage (>80%) of marriages between individuals within the valley in the last four centuries and family clustering for some traits of medical interest.12 However, the extent to which the Val Borbera population is a genetic isolate is unknown. Furthermore, it is not clear to which extent the valley’s seven villages can be considered as a single population or whether they form distinct units of a meta-population. In this study we used nuclear and mitochondrial DNA (mtDNA) data to explore the extent of genetic variation of the valley and to investigate population structure. Implications of results for gene-mapping studies are discussed.
Subjects and methods
Samples
A total of 1800 healthy individuals, spanning 18–102 years of age, gave informed consent to participate in genetic analyses. Birth, marriage and death records from the 16th century onwards have been collected and used to reconstruct pedigrees from which a pedigree-based kinship coefficient was calculated.12, 13 Data collection and genotyping of the cohort was approved by the institutional ethical committee of the San Raffaele Hospital in Milan and by the Regione Piemonte.
We used kinship information to exclude all individuals related as first-cousin or more using a custom algorithm that implements recursive removal on the basis of kinship information. We felt confident in using pedigree kinship as it has been shown to be highly correlated with genomic one.13 After removal of close relatives we classified individuals according to two criteria: (i) the four grandparents were resident in any one of the villages in the valley; and (ii) all four grandparents were resident in the same village. As detailed in Table 1, the first criterion allow us to select 267 individuals to form the ‘valley’ sample, while according to the last criterion we selected single-village samples. One of the seven villages (ROC) was excluded from the analyses because of a very small sample size (Table 1).
For comparison we added to our analyses genetic data from other reference populations (Table 1). We downloaded nuclear genotype data relative to the three populations in the HapMap collection (The International HapMap Project, Release 27, NCBI build 36). Two samples are of European origin, namely CEU (Utah residents with Northern and Western European ancestry) and TSI (Tuscans). The third sample is YRI (Yoruba) from Nigeria, Africa. We removed from CEU samples presenting cryptic relatedness as previously described.14 Finally, we added a fourth reference population, consisting of a cohort from Veneto Region, (North-East of Italy) with no apparent history of geographical isolation.15
For the mtDNA analyses, we referred to two Piedmontese populations in close geographical proximity to the Val Borbera (Table 1), namely Trino Vercellese and Val di Susa, whose 76 mtDNA control-region data are reported here for the first time. Finally, we also included published mtDNA control-region data from the Saami, a northern population known to be a genetic outlier among Europeans.16
Analyses of nuclear data
Data sets of genotypic calls at single-nucleotide polymorphisms (SNPs) were available for both the valley study cohort that was genotyped with the Illumina (San Diego, CA, USA) 370k-Quad CNV array, and the genomic reference17, 18 populations. All the data sets were filtered to retain variants that satisfy the following criteria: (a) MAF≥0.01; (b) genotype call rate >97% for markers with minor allele frequency (MAF) above 5% and genotype call rate >99% when 1%<MAF<5%; (c) Hardy–Weinberg equilibrium (HWE) P-value>0.00001. In the Valley sample HWE was calculated in a subset of individuals with probability of identity by descent >0.185. Merging of all cohorts led to overlapping 168 542 genome-wide SNPs that were used for subsequent analyses.
Pairwise genetic distance
Allele frequency differentiation (FST) between pairs of populations was calculated at each locus as σ2/π (1−π), where π is the mean allele frequency and σ2 the variance.19 Allele frequencies were estimated by allele counting in the valley sample.
Analysis of population structure
Population structure analysis was performed by means of Principal Component Analysis (PCA) 20 and genetic clustering.21 As both methods assume markers to be independent, we pruned from the genome-wide SNP set all SNPs in high LD (defined by r2≥0.4 in the valley) using MASEL,22 leaving 25 696 SNPs. This SNP number is appropriate for PCA to reach significance. Indeed, even the smallest FST (0.007 between VER and TSI, Table 2), is one order magnitude above the threshold value of 0.001 that we calculated as 1/sqrt (25 696*40), where 25 696 is the number of markers and 40 is the minimum number of chromosomes considered.20
We used PCA to summarise SNP genotype information at the level of each individual, with the aim to explore the relationships between individuals within populations and between populations. PCA was performed using Eigenstrat.23 For each component we calculated formal P-values for the presence of population substructure according to the Tracy–Widom (TW) distribution (with β=1) as described in Patterson et al.20 In order to avoid bias owing to unequal population sizes,24 we randomly sampled 88 and 20 individuals from each population when considering the valley and the villages, respectively. We had only two exceptions for ROA and ALB for which we only had available 13 and 19 individuals, respectively.
Model-based clustering analysis tests the presence of different clusters (K) in a meta-population. We applied unsupervised (ie, without prior information) clustering analysis to the whole-sample set, exploring the hypotheses of K=1 to 10 clusters using ADMIXTURE.21 Cross-validation errors for each hypothesis were calculated as described in Alexander et al.21
Run of homozygosity (ROH)
For the ROH analysis we randomly sampled 84 and 36 individuals from each population when considering the valley and the villages respectively. Similar sample size was used for the other reference populations. Genotypic data were analysed with the PLINK package version 1.0725 under default settings (ie, sliding windows 5 Mb, minimum 50 SNPs, one heterozygous genotype and five missing calls allowed). Each SNP is considered to be part of a homozygous segment when the proportion of overlapping homozygous windows is above 5%. ROHs were defined as stretches of at least 0.5 Mb with at least 25 homozygous SNPs (maximum pairwise distance=100 Kb).
LD calculation and estimate of effective population size from nuclear data
Pairwise LD was calculated using the squared correlation (r2) in genotype frequencies between 49 353 autosomal SNPs from six random chosen chromosomes (1, 3, 7, 10, 18 and 22) using PLINK.25 For all populations we estimated Ne from LD.26, 27, 28 Indeed, the expected LD value at a certain recombination distance (c) is inversely proportional to Ne and to c itself, and thus it is possible to derive Ne from LD values given that the recombination distance between the loci is known. Furthermore, recombination distance between markers is inversely proportional to the number of generations through which markers have been inherited together according to the formula t≈1/2c,28 and thus estimates of Ne at different times are possible taking into account different classes of recombination distances. One of the limitations of this approach is that the extent of recombination intervals that can be taken into account depends on the sample size, as within bins r2 is adjusted for the size of the sample used to calculate LD (r2=r2−1/n, n=sample size). Therefore, meaningless negative estimates of Ne are produced when r2 is lower than 1/n.26
In all populations, pairwise LD values separated by genetic distances comprised between 0.0625 and 0.35 cM were binned into distance categories and their average r2 was considered. This range of genetic distance offers a view of time from 20 000 to 3500 years ago (y.a.) considering 25 years generation time.29 For populations in the valley, because of highest levels of LD over recombination distances it has been possible to further extend calculations between 0.0125 and 1.25 cM providing nuclear effective populations size estimates (nNe) until 1000 y.a. Confidence intervals around estimates were derived considering chromosomes as replicates.
Analyses of mtDNA sequence data
We sequenced and analysed 360 bp (HVS-I, from np 16 024–16 383) of the mtDNA-control region (Supplementary Table 1). Sequencing was performed as previously reported.30 Within each village we randomly selected 40 individuals for which the mtDNA sequence was available or imputable using the pedigree information. Indeed, we exploited pedigree data to infer mtDNA sequences within matrilineal pedigree segments of depth of up to five generations. We are aware that this approach ignores very recent mutations. However, the loss of information is negligible as all the villages have low effective population size and the number of generations in which we assumed no mutations occurred is never higher than five.
Estimate of effective population size from mtDNA
Changes of the mtDNA effective population size (mtNe) through time were reconstructed using the extended Bayesian skyline plot (EBSP) as implemented in the BEAST software v.1.6.231 and the Hasegawa, Kishino and Yano model of nucleotide substitution.32 EBSP is a non-parametric Bayesian-based coalescent approach that makes no assumption on the demographic model of the population.31 Each coalescent interval has its own prior mtNe distribution, which is sampled during the Monte Carlo Markov Chain (MCMC), together with the coalescent tree, the branch lengths and the evolutionary parameters.33 After the removal of the burn-in, mtNe is evaluated at some specified time point on the recorded iterations of the MCMC and then interpolated to obtain its variation through time. Length of the MCMC was set to 20 000 000 iterations with a 10% burn-in and a thinning interval of 1000 to ensure all parameters to have an effective sample size above 200. Mutation rate was set to 1.3 × 107 (roughly equivalent to that in Forster et al34) and generation time to 25 years.29 To check for convergence, each analysis was run at least twice. Input files for BEAST are available upon request.
Results
Population clustering
We calculated genetic distances between pairs of populations at all available nuclear loci (Table 2). The results show comparable genetic distances among the populations of European ancestry, with apparently slightly higher structuring within the valley (Table 2).
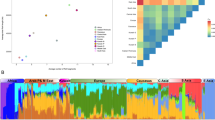
We used nuclear data to perform analyses at the individual level using both villages and valley samples. Figure 1 shows a plot of the first two principal components from randomly selected samples of equal size and after LD corrections. When considering single villages, (Figure 1b) the first two components – explaining less than 10% of the genetic variance – show significant discrimination of populations (TW P-value <0.001, Supplementary Table 2), the first one separating African from non-African populations, and the second distinguishing the valley from the other European populations. The same pattern is observed when considering the valley as a whole (Figure 1a), but only the first component is significant in this case. Model-based clustering analysis of the same data set (Figure 1c and d, Supplementary Figures 2–5) revealed K=4 as the most likely number of clusters in both cases (valley and villages, Supplementary Figure 1). Graphical representation of the proportion of ancestry in each cluster per each individual (Figure 1c and d) shows how the three main components distinguish Africans, Europeans and the valley. The fourth component that distinguishes two clusters in the MON village (Figure 1d) seems not relevant for the valley analysis, although it is the most probable one (Supplementary Figure 1).

Population clustering analyses. Model free (a, b) and model-based genetic structure analyses (c, d) relative to the valley (a, c) and the villages (b, d) samples, with respect to reference populations (see Table 1 for a list of abbreviations). In figures (a) and (b) percentages within brackets on the two axes indicate the explained variance, while P-values indicate component’s statistical significance. Both the analyses at valley and village levels show similar patterns and both clustering methods suggest poor recent genetic exchanges between the valley and the other Italians and European populations.
Long segments of autozygosity and shared haplotypes within villages with respect to other populations
ROHs are stretches of consecutive homozygous genotypic calls at adjacent SNP loci in an individual’s genome. The extent of ROHs of a genome provides a good estimate of its autozygosity at both individual and population levels. Frequent (10–13% of the genome) ROHs of short length (less than 100 kb) and less frequent ROHs of moderate length (up to 4 Mb) are expected to be found in individuals from outbred populations.35, 36, 37 Longer ROHs provide evidence for past consanguinity and population isolation.37, 38 Figure 2 presents distribution of ROHs in the different populations according to their size (in Mb). Both when considering the villages and the valley, the distribution of ROHs appears to be less left-skewed compared with other populations, suggesting a higher proportion of individuals with extended regions of autozygosity.

Statistics on the extension of the ROHs in the valley (a) and villages (b) with respect to other populations. ROHs were binned according to length and per each population the percentage of individual having at least one ROH of a given length is indicated on the y axis. As the length of the ROHs increases different trends are visible for the isolate and the reference populations. Villages behave similarly among them (b). See Table 1 for a list of abbreviations.
As a further indication of genetic isolation, we estimated the decay of LD according to recombination distance between markers. As Figure 3 shows, villages harbour the highest levels of LD compared with other populations, even for long recombination distances, similarly to other isolates.39 Interestingly, the valley sample shows an opposite trend compared with the villages, indicating that shared haplotypes tend to be longer ‘within’ villages than ‘among’ villages.

Decay of LD with increasing recombination distance measured as average r2 within recombination distance bins. As expected the population with faster decay is the YRI, whereas on the opposite villages show slower decay. The trend is inverted for the valley meta-population (dark blue) that is even faster than the YRI at recombination distances >0.15 cM.
No traces of recent expansion in populations from the villages
Using nuclear data we estimated nNe from LD. The presence of different recombination distance classes allowed us to obtain estimates at different times in a window of 20 000–1000 years ago. As Figure 4a shows, effective population size in villages is generally lower than other populations, and never exceeded 5000 individuals. Estimates for reference populations are consistent with previous analyses using the same method,26 and the trends reflect known demographic events:40, 41 a recent expansion for non-African populations and almost constant size for the African one. A similar trend is observed in the valley meta-population, whereas no signs of recent expansion can be seen in single-village samples. On the contrary, apparently a recent decline in nNe took place from 4000 years onward (Supplementary Figure 6). To further clarify this point we calculated average nNe before and after 4000 years ago for villages and the valley. As Supplementary Figure 7 shows, opposite trends took place in the villages, and the valley.

Estimates of effective population sizes. Upper graph (a) shows estimates from nuclear DNA in the last 20 000 years. Notably, villages have the lowest values of Ne and there is no signature of the recent population expansion as in the reference populations. An opposite trend is visible for the valley sample (dark blue). Finally, YRI shows a constant trend and the highest effective population size, in agreement with other estimates as mentioned in the main text. A similar trend is shown when considering estimates from mtDNA (b) data. However, given the different properties of mtDNA and the different methods we used with respect to nuclear DNA, the two estimates are not directly comparable.
We also estimated villages’ mtNe values from mtDNA (Figure 4b) and compared them with two non-isolated nearby populations of Piedmont (TRV and VDS). Two main features emerged from this comparison. First, the modern effective population size in villages is generally lower than in other populations, never exceeding 10 000. In contrast with nuclear estimates, there is higher variance among villages. The lowest mtNe is found in CAR, where mtNe is slightly above 2000, about three times smaller than Saami (a traditional isolated group 16). The second feature revealed by the EBSP analysis is a constant demography for villages, again in accordance with nuclear estimates. Conversely TRV and VDS show an increase of mtNe in the Upper Palaeolithic/Neolithic similarly to other European populations.42 Surprisingly ROA shows a different behaviour with respect to other villages. We believe this is owing to stochasticity in the reconstructed coalescent processes.
Discussion
This study shows, on the basis of several lines of evidence, that the population of the Val Borbera is a genetic isolate. First, allele frequencies summarised by PCA do not match other geographically close European and Italian populations. Our comparative analyses showed that, at the nuclear level, samples from the valley form a separate cluster from other European populations, including a northern Italian one. This indicates consistent differences in allele frequency distributions, and points to the occurrence of limited recent gene flow between them. Secondly, we observed extended regions of autozygosity with respect to other populations. In agreement with a previous study on the valley population,12 this feature indicates an excess of shared recent ancestry, suggesting that mating among recently related individuals has taken place in past generations, a condition most likely to occur during genetic isolation. Using both nuclear and mitochondrial markers we estimated a very small effective population size for the villages, suggesting a possible effect of genetic drift in reducing genetic variation within villages. Estimates of mtNe were overall greater than nuclear ones. This is some way counterintuitive when considering that mtDNA is haploid and maternally transmitted and thus should in principle be more prone to genetic drift. However, a direct comparison of the nNe and mtNe estimates is not possible as they have been produced with two different methods. Estimates of mtNe depend on knowledge of mitochondrial mutation rates and the confidence intervals of our EBSP analyses are quite large. Similarly, computing nNe from LD relies on simplifying assumptions.26 However, we are interested in the trend of population size changes through time rather than on their exact values, and thus we can be confident about our relative conclusions. Finally, contrary to other European populations, we observed a recent effective population size decline, suggesting either that the isolation is still in action or that consequences of past isolation are still present in the nuclear genome of the sampled individuals.
The second main finding of our study is that slight structuring is present among villages, within the valley. Despite clustering analysis of the villages showing no significant stratification (P-value >0.05 for both first and second principal components, Supplementary Figure 8), FST values indicate some extent of structuring, which has already been observed in isolates,43, 44 even for populations with recent shared genealogy.45 We speculate that the slight observed stratification can be related to the high proportion of marriages occurring between inhabitants of the same village, as demonstrated by analysis of marriage acts and surnames (data not shown). Further, we observe a more rapid decay of LD in the valley with respect to villages and opposite trend of LD-based estimates of nNe consistently with meta-population dynamics.46 Indeed, theoretical and simulation studies46, 47, 48, 49, 50 have demonstrated that the genealogy of lineages sampled from a deme belonging to a meta-population display a shift in the site frequency spectrum towards more intermediate frequency variants and an increase in LD compared with an unstructured population. This shift is much less pronounced when pooling lineages from more demes. This observation clearly shows how the valley is not a single panmictic unit but rather behaves as a meta-population. This finding is crucial for future gene-mapping studies, as it might help defining the unit of sampling.
Our study demonstrates that isolation took place in valley and provides insights for further gene-mapping studies. The Val Borbera population genetic and phenotypic data have been successfully used in genome-wide association meta-analyses,51, 52, 53 the first step in the identification of gene underlying complex traits in which rare gene variants are hardly identified. Isolates provide a unique opportunity to overcome this issue since rare variants frequency might be shifted towards high values. We have demonstrated that genetic drift has had a large impact on Val Borbera population and thus we expect many variants (among which some might be of relevant medical interest) that are rare in the general population to reach significant frequency values in the valley. Further the slight structuring observed might in principle allow a more fine analysis of rare frequency variants at the level of villages.
Overall, the genetic data available allowed us to investigate structure at a good resolution. However, a more accurate investigation of events that took place on a shorter time scale remain to be investigated when genomic sequence data, free of ascertainment bias, will make rare variants data available.
References
Holm H, Gudbjartsson DF, Sulem P et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet 2011; 43: 316–320.
Sulem P, Gudbjartsson DF, Walters GB et al. Identification of low-frequency variants associated with gout and serum uric acid levels. Nat Genet 2011; 43: 1127–1130.
Thorgeirsson TE, Oskarsson H, Desnica N et al. Anxiety with panic disorder linked to chromosome 9q in Iceland. Am J Hum Genet 2003; 72: 1221–1230.
Kristiansson K, Naukkarinen J, Peltonen L : Isolated populations and complex disease gene identification. Genome Biol 2008; 9: 109.
Peltonen L, Palotie A, Lange K : Use of population isolates for mapping complex traits. Nat Rev Genet 2000; 1: 182–190.
Charlesworth B : Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet 2009; 10: 195–205.
Manolio TA, Collins FS, Cox NJ et al. Finding the missing heritability of complex diseases. Nature 2009; 461: 747–753.
Service S, DeYoung J, Karayiorgou M et al. Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat Genet 2006; 38: 556–560.
Kong A, Masson G, Frigge ML et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat Genet 2008; 40: 1068–1075.
Marchini J, Howie B : Genotype imputation for genome-wide association studies. Nat Rev Genet 2010; 11: 499–511.
Palin K, Campbell H, Wright AF, Wilson JF, Durbin R : Identity-by-descent-based phasing and imputation in founder populations using graphical models. Genet Epidemiol 2011; 35: 853–860.
Traglia M, Sala C, Masciullo C et al. Heritability and demographic analyses in the large isolated population of Val Borbera suggest advantages in mapping complex traits genes. PLoS One 2009; 4: e7554.
Milani G, Masciullo C, Sala C et al. Computer-based genealogy reconstruction in founder populations. Biomed Inform 2011; 44: 997–1003.
Pemberton TJ, Wang C, Li JZ, Rosenberg NA : Inference of unexpected genetic relatedness among individuals in HapMap Phase III. Am J Hum Genet 2010; 87: 457–464.
Gambaro G, Yabarek T, Graziani MS et al. Prevalence of CKD in northeastern Italy: results of the INCIPE study and comparison with NHANES. Clin J Am Soc Nephrol 2010; 5: 1946–1953.
Tambets K, Rootsi S, Kivisild T et al. The western and eastern roots of the Saami--the story of genetic ‘outliers’ told by mitochondrial DNA and Y chromosomes. Am J Hum Genet 2004; 74: 661–682.
Altshuler DM, Gibbs RA, Peltonen L et al. Integrating common and rare genetic variation in diverse human populations. Nature 2010; 467: 52–58.
Graziani MS, Gambaro G, Mantovani L et al. Diagnostic accuracy of a reagent strip for assessing urinary albumin excretion in the general population. Nephrol Dial Transplant 2009; 24: 1490–1494.
Holsinger KE, Weir BS : Genetics in geographically structured populations: defining, estimating and interpreting F(ST). Nat Rev Genet 2009; 10: 639–650.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Alexander DH, Novembre J, Lange K : Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009; 19: 1655–1664.
Bellenguez C, Ober C, Bourgain C : Linkage analysis with dense SNP maps in isolated populations. Hum Hered 2009; 68: 87–97.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D : Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
McVean G : A genealogical interpretation of principal components analysis. PLoS Genet 2009; 5: e1000686.
Purcell S, Neale B, Todd-Brown K et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
McEvoy BP, Powell JE, Goddard ME, Visscher PM : Human population dispersal ‘Out of Africa’ estimated from linkage disequilibrium and allele frequencies of SNPs. Genome Res 2011; 21: 821–829.
Tenesa A, Navarro P, Hayes BJ et al. Recent human effective population size estimated from linkage disequilibrium. Genome Res 2007; 17: 520–526.
Hayes BJ, Visscher PM, McPartlan HC, Goddard ME : Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res 2003; 13: 635–643.
Fenner JN : Cross-cultural estimation of the human generation interval for use in genetics-based population divergence studies. Am J Phys Anthropol 2005; 128: 415–423.
Achilli A, Olivieri A, Pala M et al. Mitochondrial DNA backgrounds might modulate diabetes complications rather than T2DM as a whole. PLoS One 2011; 6: e21029.
Drummond AJ, Rambaut A : BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 2007; 7: 214.
Hasegawa M, Kishino H, Yano T : Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 1985; 22: 160–174.
Heled J, Drummond AJ : Bayesian inference of population size history from multiple loci. BMC Evol Biol 2008; 8: 289.
Forster P, Harding R, Torroni A, Bandelt HJ : Origin and evolution of Native American mtDNA variation: a reappraisal. Am J Hum Genet 1996; 59: 935–945.
Frazer KA, Ballinger DG, Cox DR et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861.
Lencz T, Lambert C, DeRosse P et al. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc Natl Acad Sci USA 2007; 104: 19942–19947.
McQuillan R, Leutenegger AL, Abdel-Rahman R et al. Runs of homozygosity in European populations. Am J Hum Genet 2008; 83: 359–372.
Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, Wilson JF : Genomic runs of homozygosity record population history and consanguinity. PLoS One 2010; 5: e13996.
Colonna V, Nutile T, Astore M et al. Campora: a young genetic isolate in South Italy. Hum Hered 2007; 64: 123–135.
Gravel S, Henn BM, Gutenkunst RN et al. Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci USA 2011; 108: 11983–11988.
Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD : Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 2009; 5: e1000695.
Soares P, Achilli A, Semino O et al. The archaeogenetics of Europe. Curr Biol 2010; 20: R174–R183.
Nelis M, Esko T, Magi R et al. Genetic structure of Europeans: a view from the North-East. PLoS One 2009; 4: e5472.
O'Dushlaine CT, Morris D, Moskvina V et al. Population structure and genome-wide patterns of variation in Ireland and Britain. Eur J Hum Genet 2010; 18: 1248–1254.
Colonna V, Nutile T, Ferrucci RR et al. Comparing population structure as inferred from genealogical versus genetic information. Eur J Hum Genet 2009; 17: 1635–1641.
Wakeley J, Aliacar N : Gene genealogies in a metapopulation. Genetics 2001; 159: 893–905.
De A, Durrett R : Stepping-stone spatial structure causes slow decay of linkage disequilibrium and shifts the site frequency spectrum. Genetics 2007; 176: 969–981.
Ray N, Currat M, Excoffier L : Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol 2003; 20: 76–86.
Stadler T, Haubold B, Merino C, Stephan W, Pfaffelhuber P : The impact of sampling schemes on the site frequency spectrum in nonequilibrium subdivided populations. Genetics 2009; 182: 205–216.
Wakeley J : Nonequilibrium migration in human history. Genetics 1999; 153: 1863–1871.
Gieger C, Radhakrishnan A, Cvejic A et al. New gene functions in megakaryopoiesis and platelet formation. Nature 2011; 480: 201–208.
Nalls MA, Couper DJ, Tanaka T et al. Multiple loci are associated with white blood cell phenotypes. PLoS Genet 2011; 7: e1002113.
Wain LV, Verwoert GC, O’Reilly PF et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet 2011; 43: 1005–1011.
Acknowledgements
This research received support from Fondazione Alma Mater Ticinensis (to AT), the Italian Ministry of the University FIRB-Futuro in Ricerca 2008 (to AA) and Progetti Ricerca Interesse Nazionale 2009 (to AA and AT), Compagnia di San Paolo, Torino, Fondazione Cariplo, Milano and Health Ministry Progetto Finalizzato (to DT). We would like to thank the inhabitants and the administrators of the Val Borbera for their kind participation in the study. A special thanks to Professor Clara Camaschella, Dr Silvia Bione, Dr Laura Crocco, Ms Maria Rosa Biglieri, Dr Diego Sabbi for help with the data collection, to Dr Gabriella Parodi, Dr Laura Gaggiano and the Cooperatova ARCA (Al) for help with the church archives. We acknowledge Professor Guido Barbujani, Professor Alberto Piazza, Professor Chris Tyler-Smith and Dr Kimmo Palin and two anonymous reviewers for valuable suggestions and comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Colonna, V., Pistis, G., Bomba, L. et al. Small effective population size and genetic homogeneity in the Val Borbera isolate. Eur J Hum Genet 21, 89–94 (2013). https://doi.org/10.1038/ejhg.2012.113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.113
Keywords
This article is cited by
-
Characterization of Danube Swabian population samples on a high-resolution genome-wide basis
BMC Genomics (2023)
-
Design and validation of a 63K genome-wide SNP-genotyping platform for caribou/reindeer (Rangifer tarandus)
BMC Genomics (2022)
-
Is there still evolution in the human population?
Biologia Futura (2022)
-
Evidence for penetrance in patients without a family history of disease: a systematic review
European Journal of Human Genetics (2020)
-
Overcoming the dichotomy between open and isolated populations using genomic data from a large European dataset
Scientific Reports (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}