
Abstract
PITX1 is a bicoid-related homeodomain transcription factor implicated in vertebrate hindlimb development. Recently, mutations in PITX1 have been associated with autosomal-dominant clubfoot. In addition, one affected individual showed a polydactyly and right-sided tibial hemimelia. We now report on PITX1 deletions in two fetuses with a high-degree polydactyly, that is, mirror-image polydactyly. Analysis of DNA from additional individuals with isolated lower-limb malformations and higher-degree polydactyly identified a third individual with long-bone deficiency and preaxial polydactyly harboring a heterozygous 35 bp deletion in PITX1. The findings demonstrate that mutations in PITX1 can cause a broad spectrum of isolated lower-limb malformations including clubfoot, deficiency of long bones, and mirror-image polydactyly.
Similar content being viewed by others


Introduction
Malformations of the limbs are common human birth defects. Genetic factors associated with these malformations often result in both upper and lower extremity anomalies.1 A basic clinical classification differentiates between reduction defects and duplications, the latter most frequently causing pre- or postaxial polydactyly.2 A rare variant of polydactyly is termed ‘mirror-image polydactyly’. This malformation is characterized by a high degree of polydactyly with a recognizable anterior/posterior axis of symmetry, that is, there is a hallux/thumb-like structure in the middle or an interdigital space with a flanking pair of digits that resemble a middle finger, index finger, thumb, toe or hallux.3 The most lateral digits on each side typically resemble fifth fingers/toes. This malformation is often observed in combination with duplication of the ulna and/or fibula or with absent radius and/or tibia (hemimelia). The condition is observed as a leading clinical sign in Laurin–Sandrow Syndrome (MIM %135750).4, 5 A translocation breakpoint in 14q13 within the designated mirror-image polydactyly 1 gene (MIPOL1) has been described in a single case of Laurin–Sandrow syndrome,6 but the genetic factors underlying mirror-image polydactyly remain largely unknown.
PITX1 encodes a bicoid-transcription factor; mutations in which have previously been reported in a large autosomal-dominant family with a clubfoot malformation.7 In this family penetrance was reduced and phenotype expression was variable, ranging from complete absence of congenital abnormalities to clubfoot, tibial hemimelia, and duplicated great toes in a single family member. This was the first indication that a broader spectrum of malformations might be due to PITX1 dysfunction. However, no other patient with tibial hemimelia was identified in this study. Recently, a second family with a deletion of the entire PITX1 gene (chr5: 134 222 383–134 463 022, NCBI36/hg18) was reported in another multi-generation family with dominantly inherited clubfoot, where no additional malformations were present.8
Here, we report heterozygous deletions of 5q31 including the PITX1 locus in two fetuses with lower-limb malformations including tibial hemimelia and mirror-image polydactyly. An intragenic deletion in PITX1 was found in a third individual with a similar phenotype of tibial hemimelia but a milder polydactyly with some mirror elements.
Methods
Subjects
Written informed consent was obtained from all the study participants after approval from the Institutional Review Boards at the participating institutions.
Sequencing
The coding exons of PITX1 (NM_002653.4) and adjacent intronic sequences were amplified and sequenced. The primer sequences are given in Supplementary Table 2. Analysis was performed with SeqPilot software (JSI Medical Systems, Kippenheim, Germany). Four hundred control chromosomes were genotyped for the c.765_799del variant using the identified mutant chromosome as positive control.
Quantitative real-time PCR (qPCR)
Genomic DNA samples were obtained from EDTA-blood. Amplicons were located within the PITX1 coding sequence and flanking regions (Supplementary Table 2). qPCR was performed as previously described.9
Fluorescence in situ hybridization (FISH)
FISH analysis was performed on metaphase chromosomes according to standard procedures. The following BAC clones were used: D5S23/D5S721 (control probes), EGR1 (detection probe) (Vysis, Abbott Park, IL, USA).
Array CGH
Total genomic DNA was purified by QIAamp DNA Blood Kit (QIAGEN, Hilden, Germany). Processing of the array (restriction digestion, fluorescent labeling, hybridization, washing, and scanning) was done in accordance with manufacturer's instructions (Agilent Oligonucleotide Array-Based CGH for Genomic DNA, Enzymatic Labelling for Blood (v6.3)). Extraction of microarray TIFF images had been done by ‘Feature Extraction’ and the following data analysis was done by the software ‘Genomic Workbench 5.0’ and ‘CGH analytics’ (both Agilent Technologies, Santa Clara, CA, USA). The following analysis settings were applied: algorithm ADM-2, filter 5 probes, log2ratio 0.29. The arrays used were: Human Genome CGH microarray kit 105A and 244K oligonucleotide array (Agilent Technologies).
Results
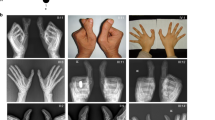
The first case we investigated was a fetus that had been diagnosed with high-degree polydactyly, hypoplasia of the corpus callosum, enlargement of the cisterna magna, and cardiomegaly. The fetus was stillborn at 34-weeks gestation after a sudden deceleration in fetal heart rate. Postmortem clinical examination revealed a small median cleft-palate, bilateral popliteal pterygia, talipes equinovarus together with mirror-image polydactyly with eight digits on each foot. The upper limbs did not show any abnormalities (Figure 1, case 1). Dysmorphic features included low-set ears, downslanting palpebral fissures, mild hypertelorism, and a flat nasal bridge. Array CGH analysis of DNA from amniotic fluid cells revealed a heterozygous 5.7 Mb deletion on chromosome 5q31.1-q31.2 (arr 5q31.1q31.2 (132 560 606–138 352 212)x1; NCBI36/hg18). This region contains 53 protein-coding genes, among them the bicoid-related homeodomain transcription factor PITX1 (Supplementary Table S1). The deletion was verified using FISH in metaphases from cultured amniocytes with a region-specific BAC probe (Figure 1 and Supplementary Figure 2) and by quantitative PCR with amplicons located in exon 2 and 3 of PITX1. The deletion was not found in parental blood samples as confirmed by array CGH and FISH suggesting a de novo occurrence.

Haploinsufficiency of PITX1 causes variable lower-limb malformations. Case 1: Reconstructions of postmortem CT scans, as well as clinical pictures showing the pes equinovarus configuration and mirror-image polydactyly. Whole body CT reconstruction shows normal upper limb and axial skeleton. FISH analysis indicating a deletion on 5q31 (arrow; green=control probe, red=locus specific probe). Case2: Ultrasound examination in a fetus in the 15th gestational week shows polydactyly and long-bone deficiency. Case 3: Note long-bone deficiency, preaxial polydactyly as well as bilateral clubfoot. The right side is more severely affected (absent tibia) than the left. Molecular genetic testing of PITX1 showed a second faster migrating amplicon of exon 3 by gel electrophoresis of the DNA from the affected individual (pat) in comparison with the maternal amplicon (co). Direct sequencing revealed a deletion of 35 bases (c.765_799del). The electropherograms show the respective genomic sequence of exon 3 (upper panel) as well as a subcloned mutated allele (lower panel) from the patient. The breakpoint is indicated by a dashed line. PITX1 is composed of three exons (E1-E3, NM_002653.4, coding region in blue). Case 1 and case 2 showed larger interstitial genomic deletions on chromosome 5 including PITX1 as determined by array CGH. The location and size of the three deletions is visualized by red bars and is given according to NCBI36/hg18.
The second case (Figure 1, case 2) had undergone ultrasound examination at 15 weeks gestation that had revealed a single lower leg bone on the left as well as mirror-image polydactyly of the right foot together with bilateral clubfoot. The upper limbs did not show any abnormalities. In this patient, array CGH analysis of amniotic fluid cell DNA showed a heterozygous deletion of 4.9 Mb on 5q31 including PITX1 (arr 5q31.1q31.2 (133 200 000–138 080 000)x1; NCBI36/hg18). The parents opted for termination of pregnancy.
To try to further elucidate the role of PITX1 in complex lower-limb malformations, eight individuals diagnosed with isolated higher degree polydactyly/mirror-image polydactyly and/or tibial hemimelia, but normally developed upper extremities underwent molecular genetic testing of PITX1. Direct sequencing of the coding exons of PITX1 as well as quantitative PCR with probes in exon 2 and 3 of the gene was performed. In one individual with bilateral preaxial polydactyly, talipes equinovarus and right tibial hemimelia we identified a heterozygous deletion of 35 bp in exon 3 of PITX1 (c.765_799del) (Figure 1, case 3, and Supplementary Figure 1). This frameshift mutation at position 256 of the 314 amino-acid protein encoded by PITX1 is predicted to cause a stop codon after incorporation of 303 additional irrelevant amino acids (p.Ala256ArgfsX303). The mutation was absent from lymphocyte DNA of the healthy mother and from 400 control chromosomes. DNA from the father was not available for molecular genetic testing. This frameshift mutation removes the C-terminal part of the protein and results in the loss of a 14 amino-acid motif, designated as OAR domain. This motif was suggested to trigger DNA binding and transactivation.10 Thus, the mutation is highly likely to be pathogenic and thus supports a role for PITX1 in complex lower-limb malformation.
Discussion
Evidence from several vertebrate species indicates that the transcription factor Pitx1 is a key molecule in limb-specific morphogenesis. Targeted disruption of Pitx1 in mice results in abnormalities of the hindlimb such as the fibula and tibia having equivalent diameters, loss of the patella and abnormalities of the calcaneous bone. This and the finding that Pitx1 is expressed only in the hindlimb suggested a role in the determination of hindlimb morphology and identity.11, 12, 13, 14 The hypothesis was supported by studies performed in chick and mouse showing that misexpression of Pitx1 in forelimbs results in the adoption of hindlimb characteristics.14, 15 In fish, pelvic loss in different natural populations of stickleback fish has occurred through regulatory mutations deleting a tissue-specific enhancer of Pitx1 and demonstrates an evolutionary change in vertebrates via this genomic region.16
In humans, a missense mutation (p.E130K) in PITX1 has been shown to segregate with dominantly inherited clubfoot with reduced penetrance in a large kindred, together with unilateral tibial hemimelia and preaxial polydactyly in one individual in this family.7 The mutation is located in the DNA-binding domain of the protein and revealed reduced transactivation in a reporter gene assay.
A second family with dominantly inherited clubfoot was recently reported to harbor a 241 kb deletion including PITX1.8 Interestingly, no signs of polydactyly or hemimelia were observed in this multi-generation family. The pathogenesis of PITX1 in clubfoot was further supported by the finding that a subset of heterozygous Pitx1 knockout mice displayed clubfoot.8
Our findings add considerable weight to the observation that PITX1 deletions can cause a spectrum of lower-limb malformations including aplasia of long bones and high-degree polydactyly. Interestingly, the CT-scan of case 1 also showed that the tibia and fibula become more alike and show a similar diameter, a feature that was also found in Pitx1−/− mice.17 The findings also extend the phenotypic spectrum to include mirror-image polydactyly as variant of polydactylies. Of note, however, full mirror-image polydactyly was only present in the fetuses with larger genomic aberrations and therefore we cannot fully exclude that other genes in the deleted genomic region might cause, or at least influence this particular phenotype. As there is increasing evidence that copy number changes in non-coding genomic elements are implicated in bone disorders,18, 19 deletions of non-coding regulatory elements upstream or downstream of PITX1 might also account for the clinical variability observed.
The digit number and identity along the anterior/posterior axis is controlled by signals from the zone of polarizing activity (ZPA) at the posterior margin of the limb bud. One of the key players in this scenario is the signaling molecule sonic hedgehog (SHH), which is expressed in the ZPA. In polydactylies, misregulation of SHH has been shown to be a crucial factor as demonstrated by mutations in the ZPA regulatory sequence of SHH, a non-coding regulatory sequence element located in 1 Mb distance of the SHH gene.20 Copy number variations and point mutations in this regulatory region have been associated with a broad spectrum of polydactylies.19 This overlapping clinical phenotype suggests an interplay between the Hedgehog pathway and PITX1 in humans.
However, the situation might be different between men and mice. Pitx1−/− mice do not show a polydactyly indicating that Pitx1 does not interfere to a significant degree with Shh signaling in the mouse. This hypothesis is corroborated by Shh expression analysis showing normal patterns of Shh expression in the ZPA of Pitx1−/− mice.12 Only a combined inactivation of Pitx1 and Pitx2 resulted in abnormal Shh expression and Pitx1 overexpression in the forelimb caused downregulation of Shh,14 indicating some degree of interaction between the pathways. A similar discrepancy between mouse and human phenotypes has been observed for WNT7A. In humans, mutations in WNT7A result in severe limb truncations whereas inactivation in the mouse causes defects in dorsal-ventral patterning.21
In summary, our findings further define the role of PITX1 in human limb development and illustrate that a spectrum of malformations is associated with PITX1 haploinsufficiency.
References
Kornak U, Mundlos S : Genetic disorders of the skeleton: a developmental approach. Am J Hum Genet 2003; 73: 447–474.
Stricker S, Mundlos S : Mechanisms of digit formation: human malformation syndromes tell the story. Dev Dyn 2011; 240: 990–1004.
Stevenson RE, Judith G H : Human Malformations and Related Anomalies. New York: Oxford University Press, 2005.
Marino-Enriquez A, Lapunzina P, Omenaca F, Morales C, Rodriguez JI : Laurin-Sandrow syndrome: review and redefinition. Am J Med Genet A 2008; 146A: 2557–2565.
Verghese R, Shah H, Rebello G, Joseph B : Pre-axial mirror polydactyly associated with tibial deficiency: a study of the patterns of skeletal anomalies of the foot and leg. J Child Orthop 2007; 1: 49–54.
Kondoh S, Sugawara H, Harada N et al: A novel gene is disrupted at a 14q13 breakpoint of t(2;14) in a patient with mirror-image polydactyly of hands and feet. J Hum Genet 2002; 47: 136–139.
Gurnett CA, Alaee F, Kruse LM et al: Asymmetric lower-limb malformations in individuals with homeobox PITX1 gene mutation. Am J Hum Genet 2008; 83: 616–622.
Alvarado DM, McCall K, Aferol H et al: Pitx1 haploinsufficiency causes clubfoot in humans and a clubfoot-like phenotype in mice. Hum Mol Genet 2011; 20: 3943–3952.
Klopocki E, Ott CE, Benatar N, Ullmann R, Mundlos S, Lehmann K : A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. J Med Genet 2008; 45: 370–375.
Semina EV, Reiter R, Leysens NJ et al: Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet 1996; 14: 392–399.
Szeto DP, Rodriguez-Esteban C, Ryan AK et al: Role of the Bicoid-related homeodomain factor Pitx1 in specifying hindlimb morphogenesis and pituitary development. Genes Dev 1999; 13: 484–494.
Marcil A, Dumontier E, Chamberland M, Camper SA, Drouin J : Pitx1 and Pitx2 are required for development of hindlimb buds. Development 2003; 130: 45–55.
Minguillon C, Del Buono J, Logan MP : Tbx5 and Tbx4 are not sufficient to determine limb-specific morphologies but have common roles in initiating limb outgrowth. Dev Cell 2005; 8: 75–84.
DeLaurier A, Schweitzer R, Logan M : Pitx1 determines the morphology of muscle, tendon, and bones of the hindlimb. Dev Biol 2006; 299: 22–34.
Logan M, Tabin CJ : Role of Pitx1 upstream of Tbx4 in specification of hindlimb identity. Science 1999; 283: 1736–1739.
Chan YF, Marks ME, Jones FC et al: Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 2010; 327: 302–305.
Lanctot C, Moreau A, Chamberland M, Tremblay ML, Drouin J : Hindlimb patterning and mandible development require the Ptx1 gene. Development 1999; 126: 1805–1810.
Kurth I, Klopocki E, Stricker S et al: Duplications of noncoding elements 5′ of SOX9 are associated with brachydactyly-anonychia. Nat Genet 2009; 41: 862–863.
Klopocki E, Mundlos S : Copy-number variations, noncoding sequences, and human phenotypes. Annu Rev Genomics Hum Genet 2011; 12: 53–72.
Hill RE : How to make a zone of polarizing activity: insights into limb development via the abnormality preaxial polydactyly. Dev Growth Differ 2007; 49: 439–448.
Woods CG, Stricker S, Seemann P et al: Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am J Hum Genet 2006; 79: 402–408.
Acknowledgements
We thank the families for their cooperation and participation in this study. This study was supported by a grant to IK by the Fritz-Thyssen-Stiftung and to EK and SM by the Deutsche Forschungsgemeinschaft.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Klopocki, E., Kähler, C., Foulds, N. et al. Deletions in PITX1 cause a spectrum of lower-limb malformations including mirror-image polydactyly. Eur J Hum Genet 20, 705–708 (2012). https://doi.org/10.1038/ejhg.2011.264
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.264
Keywords
This article is cited by
-
KAT6A mutations in Arboleda-Tham syndrome drive epigenetic regulation of posterior HOXC cluster
Human Genetics (2023)
-
Evolutionary genetics of flipper forelimb and hindlimb loss from limb development-related genes in cetaceans
BMC Genomics (2022)
-
The molecular genetics of human appendicular skeleton
Molecular Genetics and Genomics (2022)
-
Strigea robusta causes polydactyly and severe forms of Rostand’s anomaly P in water frogs
Parasites & Vectors (2020)
-
A simple strategy for heritable chromosomal deletions in zebrafish via the combinatorial action of targeting nucleases
Genome Biology (2013)
