
Abstract
Indian hedgehog (Ihh) signaling is a major determinant of various processes during embryonic development and has a pivotal role in embryonic skeletal development. A specific spatial and temporal expression of Ihh within the developing limb buds is essential for accurate digit outgrowth and correct digit number. Although missense mutations in IHH cause brachydactyly type A1, small tandem duplications involving the IHH locus have recently been described in patients with mild syndactyly and craniosynostosis. In contrast, a ∼600-kb deletion 5′ of IHH in the doublefoot mouse mutant (Dbf) leads to severe polydactyly without craniosynostosis, but with craniofacial dysmorphism. We now present a patient resembling acrocallosal syndrome (ACS) with extensive polysyndactyly of the hands and feet, craniofacial abnormalities including macrocephaly, agenesis of the corpus callosum, dysplastic and low-set ears, severe hypertelorism and profound psychomotor delay. Single-nucleotide polymorphism (SNP) array copy number analysis identified a ∼900-kb duplication of the IHH locus, which was confirmed by an independent quantitative method. A fetus from a second pregnancy of the mother by a different spouse showed similar craniofacial and limb malformations and the same duplication of the IHH-locus. We defined the exact breakpoints and showed that the duplications are identical tandem duplications in both sibs. No copy number changes were observed in the healthy mother. To our knowledge, this is the first report of a human phenotype similar to the Dbf mutant and strikingly overlapping with ACS that is caused by a copy number variation involving the IHH locus on chromosome 2q35.
Similar content being viewed by others


Introduction
Vertebrate limb development is a complex process involving many signaling pathways whose highly regulated interplay in time and space secures the formation of normal extremities. So far, many functional models of vertebrate limb development have been proposed,1 presenting evidence for several important key factors, such as Fgf, Wnt and Shh signaling. However, the exact role of Indian hedgehog (Ihh) in this intricate system is still not fully understood. Ihh signaling is known to promote endochondral bone growth through positive regulation of chondrocyte proliferation and osteoblast differentiation2, 3 and has been shown to regulate elongation of digit primordia.4 In line with this function, human mutations in IHH cause brachydactyly type 1A.5 Although Ihh signaling has been correlated with digit growth, various forms of polydactyly have been mainly associated with altered expression of Sonic hedgehog (Shh).6, 7 It has been suggested that the interaction between Shh and Gli3 defines digit number and identity.8 Conversely, the polydactylous phenotype of the mouse mutant doublefoot is caused by a deletion in cis to Ihh that leads to ectopic Ihh expression in the developing limb bud mesenchyme.9 Very recently, Klopocki et al10 associated copy number variations (CNVs) of the IHH locus with syndactyly and craniosynostosis, suggesting dysregulation of IHH through alteration of long-range regulation as the underlying mechanism for this phenotype. We here present two half-siblings in a Turkish family with a phenotype resembling acrocallosal syndrome (ACS) (MIM 200990) caused by a ∼900-kb duplication of the IHH locus indicating the importance of this locus during limb and brain development.
Methods
Patients
The family was referred to Istanbul Medical Faculty, Department of Medical Genetics, Istanbul University, Turkey, for syndrome classification and genetic counseling. Clinical evaluations were performed at different ages for the index patient (K1549). The fetus from the mother's second pregnancy (K1552) was externally examined postmortem after medical termination of the pregnancy at 18 weeks of gestation, with permission of the parents. Autopsy and cranial magnetic resonance imaging (MRI) were not performed, because the parents did not give their consent. Written informed consent was obtained for molecular studies, and genomic DNA was isolated from peripheral whole blood samples collected from the siblings K1549 and K1552, their mother (K1548) and the father of patient (K1552). The father of K1549 was not available for this study.
Single-nucleotide polymorphism (SNP) array copy number analysis
The Affymetrix genome-wide Human SNP Array 6.0. utilizing more than 906 600 SNPs and more than 946 000 probes for the detection of CNVs was used in both siblings (K1549, K1552) and their mother (K1548). Quantitative data analysis was performed with GTC 3.0.1 (Affymetrix Genotyping Console) using a reference file of ATLAS Biolabs GmbH (100 samples). We used the Segment Reporting Tool to locate segments with copy number changes in the copy number data with the assumption of a minimum of 10 kb per segment and minimum genomic size of five markers of a segment.
Quantitative real-time PCR
We used a qPCR technique for confirmation and segregation analysis of the detected duplications of the IHH locus in the two siblings (K1549 and K1552), their mother (K1548) and the father of K1552. The qPCR technique and the calculation method were described before.6 The used TaqMan assay is based on amplification and quantification of IHH in relation to an internal reference gene, albumin (ALB), in a multiplex PCR using ABIPRISM7500 (Applied Biosystems, Foster City, CA, USA). The primers and probes for IHH (marked with FAM) and ALB (marked with VIC) were designed using the PRIMER EXPRESS software (Applied Biosystems) and were purchased from Applied Biosystems and Metabion (Martinsried, Germany) (for primer sequences see Supplementary Table 1). Each reaction had 20 μl volume containing 10 μl of 2 × genotyping universal master mix buffer, 700 nM of the IHH_E1/E3 and ALB primers and 400 nM of both IHH_E1/E3 and ALB probes. 10 ng of genomic DNA were used as templates. For detection of quantitative changes in DNA amounts we used 5, 10, 15 and 20 ng of DNA in separate replicates. Thermal cycling was performed for 2 min at 50 °C, 10 min at 95 °C and then 40 cycles for 15 seconds at 95 °C and for 1 min at 60 °C. The threshold cycle parameter (Ct) was defined as the point at which the amplification plot – representing the fluorescence generated by cleavage of the probe as a function of the cycle number – passed a fixed threshold above baseline. Each replicate was normalized to ALB to obtain a ΔCt (VIC dye Ct – FAM dye Ct). To imitate the ΔCt created by one, two, three and four copies of the IHH gene, a control sample with different DNA amounts was used (VIC dye Ct (10 ng)−FAM dye Ct (5 ng)=ΔCt of one copy; VIC dye Ct (10 ng)−FAM dye Ct (10 ng)=ΔCt of two copies; VIC dye Ct (10 ng)−FAM dye Ct (15 ng)=ΔCt of three copies; VIC dye Ct (10 ng)−FAM dye Ct (20 ng)=ΔCt of four copies). All samples were then normalized to the calibrator to determine ΔΔCt (ΔCt of each sample−ΔCt of two copies).
Breakpoint analysis
We used a long-range PCR strategy to amplify the region containing the putative breakpoint. We designed several primers in inversed orientation in the region likely to contain the breakpoint as previously identified by SNP Array. We were able to amplify a single fragment of ∼6 kb (5′-primer: CAG TAA TGG AGG CTG GCA AG; 3′-primer: AGC CGG AGC AGA AGT ATG TG). Subsequently, the fragment was completely sequenced by standard Sanger sequencing.
Screening for point mutations and structural aberrations in GLI3
DNA of the index patient was screened for point mutations in the GLI3 gene using direct exon sequencing of all 15 exons, including the 14 coding exons and their adjacent intron sequences. To exclude partial or total deletions or duplications of GLI3, a quantitative PCR-based assay was designed (method analogous to the qPCR method described above).
Results
Clinical presentation of the phenotype in the Turkish family
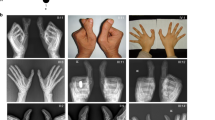
The propositus (K1549) was the first-born child to a healthy, 29-year-old mother and a 34-year-old, unrelated father. He was delivered at term by cesarean section due to breech presentation. At the age of five days he was referred to our clinic. Birthweight was 4650 g (90–97th percentile), length 56 cm (97th percentile) and OFC 39.5 cm (97th percentile). Facial features included a broad and prominent forehead, widely open anterior fontanel, hypertelorism, bilateral epicanthic folds, down slanting palpebral fissures, depressed and broad nasal root, short nose with anteverted nostrils, long philtrum, an open mouth, small mandible, dysplastic, low-set and posterior rotated small ears with underdeveloped upper helices, and auricular pits, three on the left and two on the right (Figures 1a and b).

Phenotypical characteristics of patients K1549 and K1552. (a, b) Craniofacial abnormalities of the index patient as an infant, note the broad forehead, hypertelorism, epicanthic folds, down slanting palpebral fissures, depressed and broad nasal root, short nose with anteverted nostrils, long philtrum and dysplastic, low-set and posterior rotated small ears; (c) patient at 11 years of age; (d) and (e) brain MRI of index patient; (f–h) hand and foot malformations of the index patient, note the polydactyly and cutaneous syndactyly; (i–k) radiographs of hand and foot malformations of the index patient; (l–n) female fetus (K1552) showing similar clinical features as seen in the index patient.
He had 30 fingers and toes altogether. Seven digits were present on each hand. Mirror configuration of the fingers and complete cutaneous syndactyly between the first five digits on the right and the first six digits on the left was giving a rose-bud appearance to the hands. The 7th digits were pedunculated postminimi attached to the ulnar side of the last digits (Figures 1f and g). Eight toes were present on each foot. On the right foot, there was partial cutaneous syndactyly between the first four toes and complete syndactyly between toes 5 to 7, whereas the 8th toe was apart. On the left, there was complete cutaneous syndactyly between the first seven toes, only the 8th toe was apart (Figure 1h), outwardly, the polydactyly appeared to be postaxial on the feet. Radiographs of the hands showed six well-formed metacarpals and phalanges, radiographs of the feet revealed eight metatarsals and phalanges (Figures 1i–k). Two ossification centers were observed at the distal end of the first ray metacarpal and metatarsal bones, which might argue for pre-axial polydactyly. All fingers and toes had well-formed nails. External genitalia were normal male with an undescended left testicle.
Cerebral MRI showed total agenesis of the corpus callosum, cerebral atrophy with widened inner and outer cerebral fluid interspaces and multiple cerebral cysts (Figures 1d and e). Cardiac echocardiography and EEG were normal. There was no hearing impairment.
The patient was clinically diagnosed with acrocallosal-like syndrome and was reexamined at the age of 8 and 11 years (Figure 1c). He walked independently at 36 months. At 8 years his weight was 30.5 kg (75–90th centile), height was 134 cm (25–50th centile) and OFC was 57 cm (97th centile). There was neither speech acquisition nor toilet training. He had several operations for reconstruction of the polysyndactylies. He developed severe myopia (−12 dpt.) and required surgery for glaucoma.
The half-sister of patient 1 (K1552) was the mother's second pregnancy, by a different, unrelated spouse. This pregnancy was medically terminated at 18 weeks of gestation because of the presence of similar abnormalities on antenatal ultrasound. The fetus was examined and a DNA sample was obtained, but the father did not give permission for an autopsy or cerebral MRI. Physical examination revealed that the female fetus had the same features as patient 1 (Figure 1l). All cranial sutures were widely open and a huge cystic hygroma of the neck was observed (Figure 1n). Hypertelorism, micrognathia, a small nose, and low-set deformed ears were noted (Figures 1l and n). There were seven digits on each hand, the 7th fingers being postminimi. Complete cutaneous syndactyly was present between the first five fingers, forming a rose-bud appearance of the hands (Figure 1m), the 6th and 7th fingers were apart. Both feet had eight toes, joined by complete cutaneous syndactyly. A cerebral ultrasound was uninformative. The mother of the two affected half-sibs was healthy and did not show even minor symptoms of ACS.
Identification of IHH locus duplications
Initially, we screened GLI3 as a highly relevant candidate gene for the ACS phenotype in the given family. Analysis of all 15 exons of GLI3 in the index patient provided no evidence for point mutations, deletions or duplications of GLI3. Subsequently, we performed a SNP array 6.0 in both affected siblings and the healthy mother. As paternity was different for both cases, we did not include the fathers at this stage of analysis.
The quantitative data analysis identified a significant increase in copy number reflecting three copies on chromosome 2q35 in both siblings, but no structural aberration in their mother. Patient K1549 carried a duplication of ∼900 kb in size (CN_840813 – CN_845306, position 219 584 692–220 510 875 bp, according to the hg18 assembly at the UCSC Genome Browser) including the complete IHH gene at the 5′ end of the duplication (Figure 2). Apart from IHH, 29 additional genes are located within the duplicated region. The fetus K1552 showed a similar duplication of ∼900 kb (CN_840813 – SNP_A-8363810, position 219 584 692–220 495 218 bp) with a likely overlapping 5′ breakpoint within the CCDC108 gene and a slightly different 3′ breakpoint varying by only 5 markers compared with the index patient (Figure 2). TaqMan analysis confirmed the IHH duplication in both siblings while neither their mother nor the father of K1552 showed any quantitative changes of IHH (Figure 3). Breakpoint analysis by long-range PCRs and subsequent Sanger sequencing of an amplified ∼6 kb fragment determined the breakpoints at positions 219 583 780 (5′) and 220 497 443 (3′), respectively (according to the hg 18 assembly at the UCSC Genome browser), and proved that the duplication was tandem and identical in both cases (Figure 4). The exact size of the duplication is 913 663 bp.

Genomic overview of the duplicated region on chromosome 2q35. (a) Duplications from our study and as published by Klopocki et al10 are shown in blue, the doublefoot deletion is depicted in pink. The translocation breakpoint in NHEJ1 described by Cantagrel et al17 is indicated by a green arrow. Genes within the duplicated regions are marked in yellow. The position of IHH is highlighted in red. The putative limb enhancer on chromosome 2q35 is shown in turquoise. Genome positions are given according to hg18 assembly at the UCSC Genome Browser; (b) results of quantitative SNP analysis for patients K1549 and K1552. K1549 and K1552 have a large amplification (CN-State 3) on chromosome 2 with a size of ∼900 kb. The x axis indicates the physical position of the chromosome 2q35. The y axis indicates the log2 ratio of each analyzed SNP in the region. The color reproduction of this figure is available at the European Journal of Human Genetics journal online.

CNV analysis of the IHH locus. Positions 1–4 correspond to the analyzed family (black), patients K1549 and K1552 show values representing three copies of IHH. Positions 5–16 show examples of the control cohort (gray), 17–20 show normalized standard values of a control DNA with different amounts (5 ng, 10 ng, 15 ng and 20 ng, respectively; light blue). Positions 21–24 represent the theoretically expected values of one, two, three and four IHH gene copies (dark blue). The color reproduction of this figure is available at the European Journal of Human Genetics journal online.

Illustration of identified tandem duplications. Schematic overview of genomic position of the analyzed tandem duplication. The duplicated segment is depicted in dark blue, the duplication is shown in dotted blue. Chromatograms of K1549 and K1552 show the identical breakpoints proving tandem orientation of the duplication. The break point is designated by a black line. The genomic positions of the 5′ and 3′ break points are given, and the corresponding bases are indicated by arrows. The 5′ breakpoint is located within exon 25 of CCDC108 (schematic presentation on top) and the intact and the partially duplicated copy of CCDC108 are displayed. The color reproduction of this figure is available at the European Journal of Human Genetics journal online.
Discussion
We describe a patient with craniofacial malformations, severe polysyndactyly and mental retardation who was clinically diagnosed with acrocallosal-like syndrome. Interestingly, the fetus of a second pregnancy of the mother from a different partner presented with the same phenotype. The identified duplications of the IHH locus in the affected siblings were not found in the healthy mother, which, together with the apparently dominant inheritance pattern, argues for a likely gonadal mosaicism. It has been shown in humans that inversions of the MAPT locus can predispose to microdeletions and microduplications on chromosome 17q21.31, leading to a phenotype comprising mental retardation, organ malformations and facial dysmorphism.11, 12 A predisposing structural variant, for example, an inversion on chromosome 2q35, in the mother, leading to a recurrent chromosomal rearrangement in her offspring has been considered in our cases. However, this seems unlikely as no repetitive elements or shared homologous sequences could be found within DNA flanking the duplication. Moreover, the identical breakpoints in the two sibs and the tandem orientation of the duplication support the idea of gonadal mosaicism.
The duplicated region on chromosome 2q35 contains 30 genes (including IHH; Figure 2), with a proximal breakpoint lying within the CCDC108 gene (Figure 4). CCDC108 is a putative ciliary protein, and although a ciliary function has not been ascertained for CCDC108, it has been ascribed to many other members of the CCDC gene family, like CCDC39, CCDC40 and others.13 As yet no human phenotype has been depicted for alterations in CCDC108. Seen as the 5′ breakpoint of these tandem duplications lies within CCDC108, it appears an unlikely candidate to contribute to the phenotype in our cases, because gene dosage of this gene would be expected to be normal. Most of the other genes within the duplicated region encode proteins of unknown function, and they have not yet been associated with any known human phenotype. Only a small number of genes located within the duplicated region have yet been linked to developmental phenotypes. For example, autosomal recessive mutations in OBSL1 cause 3M syndrome, a well delineated growth retardation syndrome that is irreconcilable with the phenotype described here as patients show different skeletal findings and normal intelligence.14 CHPF encodes the chondroitin polymerizing factor, a protein that is involved in chondroitin synthesis during embryonic development. RNAi knockdown of the C. elegans orthologue PAR2.4 in early embryonic stages has proven to be lethal in C. elegans.15 So far, no human phenotype associated with alterations in CHPF is known. Mutations in NHEJ1 have been associated with a Nijmegen breakage syndrome-like phenotype.16 Moreover Cantagrel et al17 described a patient with hydrocephalus, polymicrogyria and syndactyly of the fingers and toes, who carried a balanced translocation t(2;7)(q36;p22), the breakpoint on chromosome 2 disrupting NHEJ1, a gene also located within the duplicated region in our cases.17 Seen as the mechanism underlying these phenotypes is rather loss-of-function, we consider NHEJ1 unlikely to be primarily involved in the phenotype seen in this study. We conclude that the duplication's effect on IHH is most likely the main phenotypic determinant in our cases, but we cannot exclude an additive or modifying influence of other genes located in the duplications.
We searched the DECIPHER database (http://decipher.sanger.ac.uk/) to make sure that the duplication found in this study is not a common CNV. Apart from the patients described by Klopocki et al,10 only one patient is annotated in the database with a duplication of this region. This duplication encompasses 11.49 Mb, and contains 90 genes. The phenotype described is a combination of eczema, genu valgum, webbed neck and mental retardation, without poly- or syndactyly. Given the size of the duplication, it is not surprising that it is associated with a completely different phenotype, probably resulting by a misregulation of various different genes within the duplicated region.
Ihh signaling has a crucial role in vertebrate limb development, and it has been demonstrated that dysregulation of Ihh leads to severe polydactyly in the doublefoot mouse mutant (Dbf).9 The phenotype of the polydactylous Dbf mouse mutant is caused by a large deletion in cis to Ihh (Figure 2) that leads to ectopic Ihh expression in the developing limb bud mesenchyme, which in turn causes altered processing of the transcription factor Gli3.9 Gli3 is known to act downstream of Ihh in endochondral bone growth.18 It has been suggested that the Dbf deletion removes a long-range repressor, thus causing Ihh overexpression. Apart from a possible dosage effect, the duplication seen in our cases might also lead to a change in IHH transcriptional regulation through a positional effect. It has been shown for hoxD genes that the distance between regulatory elements and target genes is important for correct gene regulation and that there seems to be a correlation between the duplication size and its impact on target gene regulation.19 A putative limb-specific enhancer has been described, right next to the distal breakpoint of the ∼900 kb deletion, at position 220 713 868–220 717 300 bp on chromosome 2q35 (VISTA enhancer browser, http://enhancer.lbl.gov; Figure 2). It is conceivable that large deletions and duplications in this region might impair the correct function of this enhancer through positional effects, thus leading to imprecise expression of IHH in the limb bud. Apart from the limb malformations, Dbf exhibits craniofacial abnormalities such as a broadened skull and widely open cranial sutures, and these findings also correspond to the craniofacial abnormalities seen in our cases. Therefore, the underlying mechanism in our family could be similar to the functional consequences in the Dbf mutant. Babbs et al9 showed that in Dbf mutant mice Ihh is overexpressed in the distal limb bud mesenchyme, in an area anterior of the ZPA. This ectopic Ihh expression occurred earlier in development compared with wild-type (wt) mice. Moreover, they observed a shift in the ratio of the active and repressor forms of Gli3, an important effector of the Hh pathway. They detected higher levels of Gli3R in Dbf mutant limb buds as opposed to wt limb buds. Based on these findings, the duplication in our cases might also lead to ectopic expression of Ihh and to misregulation of the Gli3R/Gli3A ratio. However, Dbf/+ mice show primarily preaxial polydactyly, whereas in our cases we observed pre- and postaxial polydactyly and syndactyly. These phenotypic differences might be explained by a different extension of Ihh misexpression in the developing limb bud, or by a modifying effect of one of the other genes within the duplicated region.
Although the phenotypic consequences of the Dbf deletion and the large duplications identified in our study seem to be quite similar, other smaller duplications recently described cause different phenotypes in humans. The patients described by Klopocki et al10 present with syndactyly and craniosynostosis; as the latter feature is rather different from Dbf, we suggest a slightly different molecular mechanism to underlie this phenotype, that is, involvement of a single regulatory element, as opposed to a possible involvement of long-range regulators, or even a combined effect of different regulatory elements within the duplication in our cases. The effect of different chromosomal rearrangements on the regulation of IHH will be an interesting topic for future experiments. A comparison of all structural alterations affecting the IHH locus, which have yet been described in mouse and human is shown in Figure 2.
ACS is a well-defined multiple congenital malformations/mental retardation syndrome. ACS patients present with a large forehead, widely open anterior fontanel, broad nasal bridge, hypertelorism, polysyndactyly, agenesis of the corpus callosum and mental retardation. Polydactyly in ACS is mainly postaxial in the upper limb and pre-/and or postaxial in the lower limb. Syndactyly occurs in a minority of ACS patients.20 Very recently, Putoux et al20 described autosomal recessive mutations in KIF7 in families with ACS. Although the pattern of inheritance in our family was clearly not recessive, the index patient met the suggested clinical diagnostic criteria for ACS, and despite the absence of hallux duplication, a common feature of ACS,20 the diagnosis of ACS in the index patient was convincing, also supported by the severe mental retardation and agenesis of the corpus callosum, which is to be found in almost all cases with ACS.21 However, the extensive syndactyly seen in our patient is a feature more frequently seen in patients with Greig cephalopolysyndactyly syndrome (GCPS, MIM 175700). ACS shows significant overlap with GCPS, and it has been noted that clinical diagnostic criteria are not always sufficient to distinguish between both disorders.22 GCPS, caused by mutations in GLI3,23, 24 comprises predominantly postaxial polydactyly of the hands and preaxial polydactyly of the feet, syndactyly and craniofacial abnormalities such as broad nasal root, macrocephaly, hydrocephalus and ear abnormalities.25 With respect to the neurological involvement, the GCPS phenotype is thought to be milder than the phenotype observed in ACS, although corpus callosum abnormalities and mental retardation have been described in some patients.22 In order to exclude a severe presentation of GCPS in our cases, we initially excluded mutations in GLI3 in the index patient. We conclude that the phenotype of the siblings described here belongs to the same clinical spectrum as ACS and GCPS.
In this respect, it is interesting to note that Gli3 is a transcription factor, which acts downstream of both Ihh and Shh. Hh signals control Gli3 processing and the balance between its activating and its repressor form in the developing limb bud.1 Kif7, a ciliary protein and regulator of Hh signaling, recessive mutations in which cause ACS, is needed to ensure normal levels of the Gli3 repressor form (Gli3R). Homozygous Kif7 mutant mouse embryos, as well as human ACS patients show a decrease in Gli3R levels, leading to ectopic derepression of Gli3 target genes and to a polydactyly phenotype.20, 26 Evidently, the overlapping phenotypes of ACS, GCPS and our cases could thus be explained by alterations of Hh signaling and its common effector Gli3. In the future, it will be interesting to identify target genes of Gli3 that have a role in digit patterning. Identification of these target genes will help to further understand the complex mechanisms of vertebrate limb development, and these genes will constitute relevant candidate genes for polysyndactyly syndromes.
References
Zeller R, López-Rios J, Zuniga A : Vertebrate limb development: moving towards integrative analysis of organogenesis. Nat Rev Genet 2009; 10: 845–858.
Karp SJ, Schipani E, St-Jaques B, Hunzelmann J, Kronenberg H, McMahon AP : Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and –independent pathways. Development 2000; 127: 543–548.
Long F, Chung UI, Ohba S, McMahon J, Kronenberg HM, McMahon AP : Ihh signaling is directly required for the osteoblast lineage in the enchondral skeleton. Development 2004; 131: 1309–1328.
Zhou J, Meng J, Guo S et al: IHH and FGF8 coregulate elongation of digit primordia. Biochem Biophys Res Commun 2007; 363: 513–518.
Gao B, Guo J, She C et al: Mutations in IHH, encoding Indian hedgehog, cause brachydactyly Type A-1. Nat Genet 2001; 28: 386–388.
Wieczorek D, Pawlik B, Li Y et al: A specific mutation in the distant sonic hedgehog (SHH) cis-regulator (ZRS) causes Werner mesomelic syndrome (WNS) while complete ZRS duplications underlie Haas type polysyndactyly and preaxial polydactyly (PPD) with or without triphalangeal thumb. Hum Mutat 2010; 31: 81–89.
Lettice LA, Hill AE, Devenney RS, Hill RE : Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum Mol Genet 2008; 17: 978–985.
Litingtung Y, Dahn RD, Li Y, Fallon JF, Chiang C : Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature 2002; 418: 979–983.
Babbs C, Furniss D, Morris-Kay GM, Wilkie AO : Polydactyly in the mouse mutant Doublefoot involves altered Gli3 processing and is caused by a large deletion in cis to indian hedgehog. Mech Dev 2008; 125: 517–526.
Klopocki E, Lohan S, Brancati F et al: Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis. Am J Hum Genet 2011; 88: 70–75.
Zody MC, Jiang Z, Fung HC et al: Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet 2008; 40: 1076–1083.
Kirchoff M, Bisgaard AM, Duno M, Hansen FJ, Schwartz M : A 17q21.31 microduplication, reciprocal to the newly described 17q21.31 microdeletion, in a girl with severe psychomotor delay and dysmorphic craniofacial features. Eur J Med Genet 2007; 50: 256–263.
McClintock TS, Glasser CE, Bose SC, Bergmann DA : Tissue expression pattern identify mouse cilia genes. Physiol genomics 2008; 32: 198–206.
Huber C, Munnich A, Cormier-Daire V : The 3M syndrome. Best Pract Res Clin Endrocrinol Metab 2011; 25: 143–151.
Itsumikawa T, Kitagawa H, Mizuguchi S et al: Nematode chondroitin polymerizing factor showing cell-/organ-specific expression is indispensable for chondroitin synthesis and embryonic cell division. J Biol Chem 2004; 279: 53755–53761.
Dutrannoy V, Demuth I, Baumann U et al: Clinical variability and novel mutations in the NHEJ1 gene in patients with a Nijmegen breakage syndrome-like phenotype. Hum Mutat 2010; 31: 1059–1068.
Cantagrel V, Lossi AM, Lisgo S et al: Truncation of NHEJ1 in a patient with polymicrogyria. Hum Mutat 2007; 28: 356–364.
Hilton MJ, Tu X, Cook J, Hu H, Long F : Ihh controls cartilage development by antagonizing GLI3, but requires additional effectors to regulate osteoblast and vascular development. Development 2005; 132: 4339–4351.
Kmita M, Fraudeau N, Hérault Y, Duboule D : Serial deletions and duplications suggest a mechanism for the collinearity of Hoxd genes in limbs. Nature 2002; 420: 445–450.
Putoux A, Thomas S, Coene KL et al: KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet 2011; 43: 601–606.
Courtens W, Vamos E, Cristophe C, Schinzel A : Acrocallosal syndrome in an Algerian boy born ro consanguineous parents: review of the literature and further delineation of the syndrome. Am J Med Genet 1997; 69: 17–22.
Johnston JJ, Olivos-Glander I, Turner J et al: Clinical and molecular delineation of the Greig cephalosyndactyly contiguous gene deletion syndrome and its distinction from acrocallosal syndrome. Am J Med Genet A 2003; 123A: 236–242.
Vortkamp A, Gessler M, Grzeschik KH : GLI3 zinc-finger gene interrupted by translocations in Greig syndrome families. Nature 1991; 352: 539–540.
Wild A, Kalff-Suske M, Vortkamp A, Bornholdt D, König R, Grzeschik KH : Point mutations in human GLI3 cause Greig syndrome. Hum Mol Genet 1997; 6: 1979–1984.
Gollop TR, Fontes LR : The Greig cephalosyndactyly syndrome: report of a family and review of the literature. Am J Med Genet 1985; 22: 59–68.
Ingham PW, McMahon AP : Hedgehog signaling: Kif7 is not that fishy after all. Curr Biol 2009; 19: R729.731.
Acknowledgements
We thank the family members for participating in this study, Dr M Kalff-Suske for molecular genetic testing of GLI3 in our patient and Karin Boss for critically reading the manuscript. This work was supported by the German Federal Ministry of Education and Research (BMBF) by grant number 01GM0880 (SKELNET) and 01GM0801 (E-RARE network CRANIRARE) to BW and the Turkish Council of Science (TUBİTAK) by grant number 108S418 (E-RARE network CRANIRARE partner 4) to HK and OU.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Yuksel-Apak, M., Bögershausen, N., Pawlik, B. et al. A large duplication involving the IHH locus mimics acrocallosal syndrome. Eur J Hum Genet 20, 639–644 (2012). https://doi.org/10.1038/ejhg.2011.250
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.250
Keywords
This article is cited by
-
Super-enhancer hijacking drives ectopic expression of hedgehog pathway ligands in meningiomas
Nature Communications (2023)
-
Deletion in 2q35 excluding the IHH gene leads to fetal severe limb anomalies and suggests a disruption of chromatin architecture
European Journal of Human Genetics (2019)
-
DB2: a probabilistic approach for accurate detection of tandem duplication breakpoints using paired-end reads
BMC Genomics (2014)
