
Abstract
Individuals with autism are more likely to carry rare inherited and de novo copy number variants (CNVs). However, further research is needed to establish which CNVs are causal and the mechanisms by which these CNVs influence autism. We examined genomic DNA of children with autism (N=41) and healthy controls (N=367) for rare CNVs using a high-resolution array comparative genomic hybridization platform. We show that individuals with autism are more likely to harbor rare CNVs as small as ∼10 kb, a threshold not previously detectable, and that CNVs in cases disproportionately affect genes involved in transcription, nervous system development, and receptor activity. We also show that a subset of genes that have known or suspected allele-specific or imprinting effects and are within rare-case CNVs may undergo loss of transcript expression. In particular, expression of CNTNAP2 and ZNF214 are decreased in probands compared with their unaffected transmitting parents. Furthermore, expression of PRODH and ARID1B, two genes affected by de novo CNVs, are decreased in probands compared with controls. These results suggest that for some genes affected by CNVs in autism, reduced transcript expression may be a mechanism of pathogenesis during neurodevelopment.
Similar content being viewed by others


Introduction
Autism is a pervasive developmental disorder characterized by a restricted, repetitive range of behaviors and interests, and challenges with social interactions and communication. Autism spectrum disorders (ASD) typically present before 3 years of age, affect as many as 1% of children, and are diagnosed in four times the number of boys as girls. Autism is highly heritable, with up to 90% concordance in monozygotic twins.1
Recent evidence suggests autism is associated with genomic structural variation both genome wide and at hotspots, with de novo and inherited CNVs, and with overrepresentation of certain genes within rare CNVs.2, 3, 4, 5, 6, 7, 8, 9, 10 Furthermore, many structural variants associated with autism overlap CNVs associated with other neuropsychiatric conditions or syndromes.11, 12, 13 These findings indicate that autism is characterized by marked genetic heterogeneity; with the involvement of many different highly-penetrant mutations across many different genes and pathways. Although the association between rare copy number mutations and autism is well established, specific causal mechanisms remain unclear, particularly in the case of CNVs transmitted by unaffected parents.
The objective of this study was to determine whether, using denser array comparative genomic hybridization (aCGH) platforms, association between autism and rare CNVs could be extended to de novo and inherited structural variants as small as 10 kb. Secondarily, we sought to identify biologically relevant changes in expression after de novo mutation or between affected probands and unaffected transmitting parents.
Materials and methods
Subjects included 41 youth with ASD and their parents.14 Of the 41 cases, 35 were male and 6 were female. Cases were predominantly European American (EA; 29/41), and also included three Asian Americans, two Hispanic and two African Americans (AA), and five subjects who failed to report ancestry. For this study, we only analyzed complete mother–father–child trios as verified by forensic mircosatellite markers. Controls were 367 individuals (319 EA and 48 AA) that were at least 30 years of age with no history of psychiatric symptoms by self-report.15
DNA from individuals with autism, their parents, and controls was hybridized to Nimblegen HD2 (Roche Nimblegen, Madison, WI, USA) microarray slides against a well-characterized reference subject, SKN1. The HD2 array features 2.1 million probes spaced at an average of 1.2 kb across the genome. aCGH data was normalized and CNVs were called using a sliding-window algorithm with a 10-probe (∼10 kb) minimum size threshold. Parent-of-origin for CNVs in autism probands was determined by probe-by-probe signal comparison of the proband and both parents across the CNV. Controls were split into two groups: 244 were used for the identification of normal variation and 123 were used to test for differences in rare CNV prevalence compared with autism cases. Rare CNVs were defined as having <30% overlap with a CNV identified in our control sample or in the Database of Genomic Variants.16 We decided in advance to use <30% in order to permit some overlap with known polymorphisms, while also stringently removing known CNVs that have different reported break points due to differences in signal strength or between aCGH platforms and detection algorithms. Enrichment of functional gene classes tested using PANTHER17 followed up by direct case versus control group comparison.
For a selected set of CNVs in regions with previous evidence of association to neurological phenotypes and of dosage-sensitivity or allele-specific effects we tested for either a novel stable transcript or a change in the expression in affected genes relative to the transmitting parents and/or wild-type unaffected subjects. In both the cases, total RNA or cDNA was prepared from transformed lymphoblasts from each trio and one healthy wild-type control. Expression levels were estimated using TaqMan expression assays run in triplicate with TBP as an internal control following the manufacturer's protocol (http://www.appliedbiosystems.com).
Results
In comparison with controls, we found enrichment in the percent of cases harboring rare genic CNVs ∼10 kb or larger (odds ratio (OR)=2.42; 95% CI: 1.07–6.10; P=0.04) (Table 1). The increased carrier frequency was accompanied by a significant increase in CNV- and gene burden in cases, as evaluated by Wilcoxon test. Odds ratios for carrying a rare or de novo CNV were greater for deletions, and increased with CNV size (Figure 1). A log-linear test for trend of OR across size classes was significant at P=3 × 10−9 for deletions, but not significant for duplications. The majority of rare CNVs identified in autism cases were inherited (133/136) based on aCGH analysis, indicating a minimal false-positive call rate for CNVs included in our analysis. The other three CNVs in cases, all deletions, were predicted to be de novo. Break points of these de novo deletions were confirmed by PCR and their presence verified in DNA of cases and their absence verified in the parents. Supplementary Table 1 contains all rare CNVs identified in cases and controls.

Size-stratified odds ratios (OR) for rare genic deletions and duplications. OR generated by comparing the number of cases and controls carrying one or more rare genic deletions or duplications of the given size class. Significance tested using χ2-test. All size classes are independently significant for deletions at P<0.05, whereas for duplications only mutations 50–200 kb show a significant OR.
Considering gene representation within rare CNVs, three functional classes were identified as enriched in cases, whereas no functional classes were enriched in controls (Supplementary Table 2). The strongest enrichment was for transcription-related functions (OR=9.73; P=0.0008) with 15% of cases harboring a deletion overlapping a gene annotated to this term versus less than 2% of controls. Other functional classes significantly enriched in case events were receptor and receptor activity (OR=7.84; P=0.00002), and nervous system development (OR=6.46; P=0.0004). Overall, 15/41 (37%) cases had at least one deleted gene annotated to one of these functional classes versus 8/123 (7%) of controls (OR=8.08; P=0.000002). Four patients (10%) carried both maternally and paternally transmitted deletions that affected enriched gene classes, thus supporting a two-hit model for some cases.18
We tested one predicted truncation and nine full-gene deletions or duplications for transcript level differences associated with rare case CNVs in genomic loci that have previous evidence supporting a link to autism (Figure 2). The specific genes we selected for expression analysis have evidence supporting dosage sensitivity or allele-specific effects, increasing the likelihood that expression changes linked to copy number variation have functional relevance. For the one predicted truncation, we detected a stable mutant transcript of ARID1B after de novo deletion. Sanger sequencing of the mutant transcript identified the deleted exons and the premature stop codon.

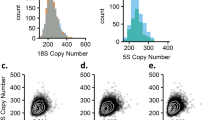
CNVs with transcript-level differences between probands and parents. For all trios shown, individual 1 is the case, 2 is the mother, and 3 is the father. The left side of each panel shows the aCGH data and the right side shows the transcript effect. Error bars on expression plots represents ±2 SD. (a) Deletion within ARID1B that results in a truncated protein missing the ARID domain and the uncharacterized and DUF3518 domain. The stable transcript was amplified from cDNA in proband, but not in parents. Trace images showing deletion break points and new stop after codon 520. (b) De novo deletion on 22q11 with decrease in relative PRODH expression shown for the deletion carrier. (c) Maternally transmitted duplication overlapping the Beckwith–Wiedemann region at 11p15. Expression of ZNF214 in the trio and a control individual showing significant stepwise decrease in expression in the unaffected mother and affected proband. (d) Maternally transmitted promoter deletion near CNTNAP2. Relative expression of CNTNAP2 in the trio and a control individual shows significant stepwise decrease in the unaffected carrier mother and affected proband.
Of the genes that were fully deleted or duplicated by rare CNVs, we observed differential expression of three genes. We found decreased PRODH expression level after de novo deletion in the proband versus wild-type parents and one control (P=0.01–0.03). We observed decreased ZNF214 expression compared with the transmitting mother (P=0.003), the wild-type father (P=0.002), and a control individual (P=0.001). Expression of the transmitting mother was also significantly lower than the father (P=0.005) and control (P=0.001). No significant trend was observed in ZNF215 expression, which may also be affected by the CNV. Similarly, a case carrying a promoter deletion exhibited decreased CNTNAP2 expression relative to the transmitting mother (P=0.0004), the wild-type father (P=0.004), and control (P=0.002). Expression in the mother was also significantly decreased relative to the father (P=0.02) and control (P=0.02). We did not see an expression effect associated with an inherited intronic deletion in GRID1. Other genes tested (GRIK2, SIM1, SNRPN, AFF2) were not expressed at detectable levels in lymphoblasts.
Discussion
Previous studies have demonstrated a clear link between rare CNVs and autism. Our analysis extends previous findings to indicate a relationship between smaller (to ∼10 kb size) rare and de novo CNVs, and demonstrates expression level phenotypes specific to affected carriers. Although heterogeneous, the affected genes identified in this study share common functions and regulatory mechanisms.
An inherent challenge to identifying rare disease-causing mutations is the lack of power required to demonstrate a statistically significant association for any given variant, as each mutations may only occur in a small number of affected individuals. However, for rare or common alleles, the ultimate proof of causality requires on the establishment of biological relevance. To this end, we demonstrate transcript level differences related to inherited and de novo CNVs.
Each of the genes for which we identified transcript level differences have evidence suggesting a potential causal relationship for autism. ARID1B mutations, such as the de novo deletion identified here, could have effects in autism similar to the affects of mutations in the related SMARC2A gene in schizophrenia.19, 20 Findings of decreased PRODH expression in autism cases carrying a deletion in this gene have been reported5 and PRODH is known to be dosage-sensitive in mouse neurodevelopment.21 Imprinting has been shown to cause allele-specific expression of ZNF215 but not the neighboring ZNF214 gene,22 and this region is associated with maternally transmitted balanced chromosomal abnormalities causing Beckwith–Wiedemann syndrome, which can be comorbid with autism.23 Described ZNF215 isoforms that contain a 3′ region that is antisense to ZNF214 are completely duplicated by the CNV reported here and these isoforms have been hypothesized to have a role in ZNF214 regulation.22 CNTNAP2 has been implicated in autism through association, linkage studies, and brain expression pattern,24, 25, 26 and there is some evidence for a parent-of-origin effect for CNTNAP2 mutations.25 Although other rare case CNVs overlapped regions with parent-of-origin effects (eg, SNRPN27 and GRIK228), the genes were not expressed in lymphoblasts, precluding expression analysis.
We report a strong enrichment in rare CNVs and specific gene classes in autism cases despite a relatively small number of trios profiled. The small number of genes and subjects tested and the use of lymphoblasts in the expression analysis are limitations of this study. Further characterization of normal variation in transcript abundance will be useful to determine whether the expression differences we report are biologically relevant. Ultimately, functional experiments such as assays for dosage-sensitive or allele-specific dominant phenotypes in neuronal cell lines are necessary. Finally, for expression analysis, we focused solely on genes with previous evidence suggesting a link to autism. There are additional case CNVs in loci associated with neurological processes and phenotypes that may be important for autism.
Although rare CNVs are overrepresented in autism, such mutations are also found in unaffected individuals. We show here that certain mutations are associated with differential expression relative to the transmitting parent. This suggests that allele-specific expression may control penetrance and explains why some carriers are unaffected. Our results indicate that screening and functional analysis combined with testing transcript-level phenotype may be an effective way to identify which rare mutations truly contribute to disease.
References
Bailey A, Le Couteur A, Gottesman I et al: Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 1995; 25: 63–77.
Autism Genome Project Consortium: Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 2007; 39: 319–328.
Christian SL, Brune CW, Sudi J et al: Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol Psychiatry 2008; 63: 1111–1117.
Glessner JT, Wang K, Cai G et al: Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009; 459: 569–573.
Guilmatre A, Dubourg C, Mosca A-L et al: Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry 2009; 66: 947–956.
Marshall C, Noor A, Vincent J et al: Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 2008; 82: 477–488.
Morrow EM, Yoo S-Y, Flavell SW et al: Identifying autism loci and genes by tracing recent shared ancestry. Science 2008; 321: 218–223.
Pinto D, Pagnamenta AT, Klei L et al: Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010; 466: 368–372.
Sebat J, Lakshmi B, Malhotra D et al: Strong association of de novo copy number mutations with autism. Science 2007; 316: 445–449.
Weiss LA, Shen Y, Korn JM et al: Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008; 358: 667–675.
Abrahams BS, Geschwind DH : Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet 2008; 9: 341–355.
Hogart A, Wu D, Lasalle JM, Schanen NC : The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis 2008; 46: 86–93.
Cook Jr EH, Scherer SW : Copy-number variations associated with neuropsychiatric conditions. Nature 2008; 455: 919–923.
King BH, Hollander E, Sikich L et al: Lack of efficacy of citalopram in children with autism spectrum disorders and high levels of repetitive behavior: citalopram ineffective in children with autism. Arch Gen Psychiatry 2009; 66: 583–590.
Talati A, Fyer AJ, Weissman MM : A comparison between screened NIMH and clinically interviewed control samples on neuroticism and extraversion. Mol Psychiatry 2008; 13: 122–130.
Iafrate AJ, Feuk L, Rivera MN et al: Detection of large-scale variation in the human genome. Nat Genet 2004; 36: 949–951.
Mi H, Guo N, Kejariwal A, Thomas PD : PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res 2007; 35: D247–D252.
Girirajan S, Rosenfeld JA, Cooper GM et al: A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet 2010; 42: 203–209.
Nagl Jr NG, Wang X, Patsialou A, Van Scoy M, Moran E : Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J 2007; 26: 752–763.
Koga M, Ishiguro H, Yazaki S et al: Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Hum Mol Genet 2009; 18: 2483–2494.
Meechan DW, Tucker ES, Maynard TM, LaMantia A-S : Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc Natl Acad Sci USA 2009; 106: 16434–16445.
Alders M, Ryan A, Hodges M et al: Disruption of a novel imprinted zinc-finger gene, ZNF215, in Beckwith-Wiedemann syndrome. Am J Hum Genet 2000; 66: 1473–1484.
Kent L, Bowdin S, Kirby GA, Cooper WN, Maher ER : Beckwith Weidemann syndrome: a behavioral phenotype-genotype study. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 1295–1297.
Alarcón M, Abrahams BS, Stone JL et al: Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet 2008; 82: 150–159.
Arking DE, Cutler DJ, Brune CW et al: A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet 2008; 82: 160–164.
Poot M, Beyer V, Schwaab I et al: Disruption of CNTNAP2 and additional structural genome changes in a boy with speech delay and autism spectrum disorder. Neurogenetics 2010; 11: 81–89.
Herzing LBK, Cook EH, Ledbetter DH : Allele-specific expression analysis by RNA-FISH demonstrates preferential maternal expression of UBE3A and imprint maintenance within 15q11- q13 duplications. Hum Mol Genet 2002; 11: 1707–1718.
Bah J, Quach H, Ebstein RP et al: Maternal transmission disequilibrium of the glutamate receptor GRIK2 in schizophrenia. Neuroreport 2004; 15: 1987–1991.
Acknowledgements
This work was supported by the National Institutes of Health grants R01MH083989, R01MH076431, R01HG004222, U54MH066398, U01HD045023, U54MH066673, U54MH068172, U54MH066418, U54MH066494, 5T32ES007032, and T32ES015459.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Supplementary information
Appendix
Appendix
Bryan H King, MD, Seattle Children's Hospital, University of Washington, Seattle, WA; Eric Hollander, MD, Institute of Clinical Neuroscience, New York, NY; Linmarie Sikich, MD University of North Carolina, Chapel Hill, NC; James T McCracken, MD, University of California at Los Angeles, Los Angeles, CA; Lawrence Scahill MSN, PhD, Yale University, New Haven, CT; Joel D Bregman, MD, North Shore–Long Island Jewish Health System, Great Neck, NY; Craig L Donnelly, MD, Dartmouth Medical School, Hanover, NH; Kimberly Dukes, PhD, Boston University, Boston, MA; Deborah Hirtz, MD, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Ann Wagner, PhD, National Institute of Mental Health, Rockville, MD.
Rights and permissions
About this article
Cite this article
Nord, A., Roeb, W., Dickel, D. et al. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur J Hum Genet 19, 727–731 (2011). https://doi.org/10.1038/ejhg.2011.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.24
Keywords
This article is cited by
-
Neurobiology of ARID1B haploinsufficiency related to neurodevelopmental and psychiatric disorders
Molecular Psychiatry (2022)
-
Early postnatal serotonin modulation prevents adult-stage deficits in Arid1b-deficient mice through synaptic transcriptional reprogramming
Nature Communications (2022)
-
Role of JAK-STAT and PPAR-Gamma Signalling Modulators in the Prevention of Autism and Neurological Dysfunctions
Molecular Neurobiology (2022)
-
Neuroanatomy and behavior in mice with a haploinsufficiency of AT-rich interactive domain 1B (ARID1B) throughout development
Molecular Autism (2021)
-
Identification of primary copy number variations reveal enrichment of Calcium, and MAPK pathways sensitizing secondary sites for autism
Egyptian Journal of Medical Human Genetics (2020)
