
Abstract
MLL2 mutations are detected in 55 to 80% of patients with Kabuki syndrome (KS). In 20 to 45% patients with KS, the genetic basis remains unknown, suggesting possible genetic heterogeneity. Here, we present the largest yet reported cohort of 116 patients with KS. We identified MLL2 variants in 74 patients, of which 47 are novel and a majority are truncating. We show that pathogenic missense mutations were commonly located in exon 48. We undertook a systematic facial KS morphology study of patients with KS at our regional dysmorphology meeting. Our data suggest that nearly all patients with typical KS facial features have pathogenic MLL2 mutations, although KS can be phenotypically variable. Furthermore, we show that MLL2 mutation-positive KS patients are more likely to have feeding problems, kidney anomalies, early breast bud development, joint dislocations and palatal malformations in comparison with MLL2 mutation-negative patients. Our work expands the mutation spectrum of MLL2 that may help in better understanding of this molecule, which is important in gene expression, epigenetic control of active chromatin states, embryonic development and cancer. Our analyses of the phenotype indicates that MLL2 mutation-positive and -negative patients differ systematically, and genetic heterogeneity of KS is not as extensive as previously suggested. Moreover, phenotypic variability of KS suggests that MLL2 testing should be considered even in atypical patients.
Similar content being viewed by others

Introduction
Kabuki syndrome (KS, Kabuki make-up syndrome or Niikawa–Kuroki syndrome) is an autosomal dominant condition that arises de novo in a majority of cases and is characterised by a recognisable facial phenotype of interrupted high-arched eyebrows, long palpebral fissures, eversion of lateral part of lower eyelids, broad depressed nasal tip, large prominent earlobes and pillowed lower lip.1, 2 All children with KS are globally delayed and have mild-to-moderate learning disability (for review of clinical features, see Adam and Hudgins3). They also have a high incidence of internal malformations, involving, but not limited to, the heart, kidneys, gastrointestinal system, skeletal system and eyes. KS is commonly associated with severe feeding difficulties, hypotonia, postnatal failure to thrive, obesity in later childhood and frequent infections. Rare problems include idiopathic thrombocytopenic purpura, seizures, autistic traits, delayed puberty and tumours.
Recently, whole exome sequencing led to the identification of mutations in MLL2 as the basis of KS in a majority of the patients.4 Still, the underlying cause cannot be identified in 20 to 45% of patients with a presumed diagnosis of KS, suggesting possible genetic heterogeneity.5, 6, 7, 8 To investigate the spectrum of mutations associated with KS, we sequenced MLL2 in 116 patients with clinically suspected KS. To test if KS is genetically heterogeneous, we systematically evaluated differences between the phenotype of patients in whom MLL2 mutations were found versus those in whom mutations were not detected.
Patients and methods
Patient ascertainment
Before the discovery of MLL2 mutations as a cause of KS, we had ascertained a highly selected group of ∼35 patients, based on clinical features and facial dysmorphology, through the Clinical Genetics clinics in Manchester. Of these 35 patients, 18 were included in the study reported by Hannibal et al.8 After the gene discovery, we expanded our cohort by accepting patients with possible KS from Clinical Genetics departments, mostly across the United Kingdom. Referring clinicians were asked to provide DNA samples from their patients and, if possible/required, from both parents. A clinical proforma was designed to gather phenotypic information from the referring clinicians. Photographs of patients were also collected wherever possible. The study was approved by the Central Manchester Research Ethics Committee (02/CM/238) and the University of Manchester Ethics Committee. Fully informed signed consents were taken from legal guardians of the patients by the referring clinicians. The study was performed in accordance with the Declaration of Helsinki protocol.
Mutation screening of MLL2 gene
All the patients were screened for mutations in the MLL2 gene. PCR amplification for all the 54 exons spanning the MLL2 gene was performed using primer sequences and the PCR conditions previously described.4 PCR amplification was carried out on Veriti thermal cycler or 2720 thermal cycler (Applied Biosystems, Paisley, UK) using the Reddy Mix Custom PCR Master Mix (ABgene, Epsom, UK, catalogue no: AB-0575/DC/LD/b) according to the manufacturer's instructions. Amplified products were cleaned using Agencourt AMPure XP (Beckman Coulter Genomics, Takeley, UK) system on an automated Beckman Coulter Liquid Handler, Biomek 3000, as per the manufacturer's instructions. The purified PCR products were then subjected to direct sequencing using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). All the sequencing reactions were cleaned using Agencourt CleanSeq on automated Beckman Coulter Liquid Handler and were then sequenced in the departmental core facility on an ABI prism 3100 Genetic Analyser (Applied Biosystems). The sequence data generated were exported and analysed graphically using the DNA sequence analysis software, STADEN (http://staden.sourceforge.net/). The changes observed were checked against the NCBI SNP database to identify known single-nucleotide polymorphisms (SNPs). Sequence variations found were reported according to the Human Genome Variation Society nomenclature. Where available, DNA from the parents of the patients with mutations was screened to investigate if the change was de novo or inherited.
All the mutations that were identified in this study and in our patients reported by Hannibal et al8 were independently confirmed. We designed new primer sets for this step to avoid amplicons of >700 base pairs. These primer sequences are available on request.
Facial KS morphology score
Clinical geneticists and dysmorphologists from Manchester, Liverpool, Leeds, Newcastle upon Tyne, Sheffield and Cardiff meet six monthly in Manchester at ‘M62 dysmorphology meeting’ to discuss unusual cases. We decided to test if facial dysmorphism can be a reliable predictor of MLL2 mutation status in our cohort. We showed 37 facial photographs of 33 patients from our cohort to 23 dysmorphologists who had gathered for the M62 meeting in June 2011. Photographs for 15 MLL2-negative patients were available. Photographs of all these 15 patients were included in the study to eliminate selection bias and provide enough statistical power. Although more photographs of patients with definite MLL2 mutation were available, only 18 were included in the study. This was done to ensure that the presentation lasted for <10 min to avoid participant boredom that could adversely affect the results. Majority of the photographs for MLL2 mutation-positive patients were selected randomly, but some with atypical appearance were included intentionally to test the whole spectrum of facial dysmorphism of KS. Participants were blind to clinical histories and mutation status of the patients. Each photograph was shown for 15 s and participants were asked to rate the likelihood of the patient having KS on an ordinal scale of one to five (one for ‘unlikely KS’, three for ‘possible KS’ and five for ‘definite KS’). The results were analysed using Mann–Whitney test. To assess if facial KS morphology scores were age dependent, we included two photographs taken at different ages for four patients.
Genotype–phenotype analysis
Detailed information on clinical features was available for 63/116 patients (40 MLL2 mutation positive and 23 MLL2 mutation negative). We concentrated on analysing differences between those features where information was reliably quantifiable and was available in majority of the cases. Therefore, we did not analyse differences between growth, developmental delay and learning disability. Two-tailed P-values were generated by Fisher's exact test.
Results
Mutation analysis
We sequenced all 54 exons and the exon–intron boundaries in 98 patients with a suspected diagnosis of KS. For the purpose of analysis, we have also included results from our additional 18 patients (giving a total of 116) who have already been reported by Hannibal et al.8 All the patients previously reported in another study have been identified clearly in the comments box of each table (Supplementary Tables 1–4).
We ascribed ‘definite pathogenicity’ only to mutations resulting in truncated protein or obvious splicing error or missense changes that were proven to be de novo in at least one patient. If both parental samples were unavailable, a missense change was considered to be ‘likely pathogenic’ if it has been identified in more than one patient and in silico predictions indicated high degree of conservation and disruption of protein function. Changes that were inherited from an unaffected parent or found concurrently in the same patient with another definitely pathogenic variant were assigned as ‘unlikely pathogenic’. Changes that could not be qualified to any of these categories were labelled to be of ‘uncertain pathogenicity’.
Following our criteria, no pathogenic variants were found in 42/116 (36.2%) patients. In 74/116 (63.8%) patients, we identified MLL2 variants, out of which 66 variants were inferred to be definite disease causing and 2 to be likely pathogenic (Figure 1). We could not be certain of the significance of six changes. Out of 74 changes, 25 were nonsense mutations, 21 were frameshift deletions, 9 were frameshift insertions, 10 were splice-site variants, 9 were missense changes and 2 were in-frame deletions. Both the changes classified as likely pathogenic were missense changes. Out of six changes with uncertain significance, two potentially affected splicing and four were missense changes. In addition, 47 changes were novel variants.

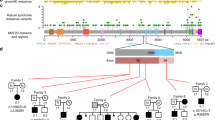
Spectrum of MLL2 mutations. Schematic representation of MLL2 mutations identified in this study. The gene structure shows all 54 coding exons and the protein structure shows the protein domains and motifs. Arrows indicate the exonic location of the mutations identified in the patients. Nonsense mutations are shown in red, frameshift in/dels are shown in green, splice-site mutations are shown in blue and missense mutations and in-frame deletions are shown in purple. Novel variants are underlined and variants of uncertain pathogenicity are highlighted by *.
Facial KS morphology scores
Mean and median values for facial KS morphology scores were calculated for each patient. We considered an average score of >3.5 and <3 as being indicative of features highly compatible or incompatible with KS, respectively. For scores between 3 and 3.5, we assumed that facial dysmorphism was not enough to strongly suggest or reject KS as a diagnosis for the individual.
Table 1 provides a summary of facial KS morphology scores in all 33 patients. Our analysis showed that 13/15 MLL2 mutation-negative patients had a mean score of <3 for facial KS morphology, whereas 12/18 MLL2 mutation-positive patients scored >3.5. Although there is some overlap between the scores of two groups, the difference between facial KS morphology scores of MLL2 mutation-positive and -negative KS patients was highly significant, with the P-value of <0.0001.
Genotype–phenotype correlation
We analysed our phenotype data to investigate if there were any differences between other clinical features of MLL2 mutation-positive and -negative groups. The results showed that feeding problems, urogenital anomalies, early breast buds, joint dislocations and cleft palate were significantly more common in patients with MLL2 mutations (Table 2).
Discussion
MLL2 is a histone H3 lysine 4 (H3K4)-specific methyl transferase that belongs to the SET1 family of human SET-domain protein methyltransferase superfamily.9, 10 It is a large protein that consists of seven plant homeodomains (PHD, for protein–protein interaction), one high-mobility group domain (for binding to DNA with low sequence specificity), five LXXLL motifs (for interaction with nuclear receptors), one each FYRC and FYRN domains (for heterodimerisation between terminal fragments of MLL) and a single SET domain (for histone lysine methylation) (Figure 1).10 MLL2 functions as a part of multiprotein complex, ASCOM, which binds to the regulatory sites of the target genes.11 It is important for epigenetic transcriptional activation, it interacts with oestrogen receptor-α and is important for embryonic development.9, 12
We identified variants in 74/116 (63.8%) patients with KS in our study (Figure 1). Ng et al4 discovered mutations in 35/53 (66%) cases of KS through a combination of exome and Sanger sequencing. Paulussen et al,5 Li et al6 and Micale et al7 identified MLL2 mutations in 34/45 (75.5%), 19/34 (55.8%) and 45/62 (72.5%) patients, respectively. Most recently, Hannibal et al8 reported 57 more patients with KS and found mutations in 46 individuals (80%), including 17 of our 18 patients. We present here the largest cohort of MLL2 mutation-positive patients with KS identified as yet. Overall mutation detection rate between different studies is variable, which is likely a reflection of differences in ascertainment.
Including this study, there are now 232 KS kindreds with MLL2 mutations in the published literature. We have analysed the MLL2 mutation spectrum by combining our data with what is already published.
Truncating mutations
Out of 232 published MLL2 mutation-positive KS patients, including 55 patients reported here, 170 (73.2%) have truncating mutations (Supplementary Table 1). A number of these mutations have now been reported in more than one patient and are highlighted in the Supplementary Table 1.
Out of 170, 86 (more than half) are nonsense mutations. In addition, 59 patients have deletions, with the majority of a single base. The biggest reported deletion is of 32 bases. Notably, c.6595delT (p.Y2199IfsX65) in exon 31 has been described in five patients, including two patients in our study. It has been shown to be de novo in three patients, confirming that it is a recurrent mutation. Also, 22 patients have small duplications or insertions. Three patients have complicated insertion and duplication, with two involving exon 34.
Of note, a number of exons are disproportionately affected by truncating mutations. For example, exon 52, which codes for 0.44% of the translated transcript, accounts for three mutations in four patients, more than five times the expected rate. Exons 27, 33, 40 and 53 also account for excessively large number of truncating mutations, although the sample size is insufficient to draw statistical conclusions.
The largest numbers of truncating mutations have been found in exon 39 (36 mutations in 39 patients) and exon 48 (16 mutations in 21 patients). Exons 39 and 48 code for 16.8 and 6.9% of MLL2 protein, correspondingly. The truncating mutation closest to N and C terminals of the protein are p.C158VfsX50 (exon 4) and p.R5501X (exon 53).
Splice-site mutations
We have added 10 splice-site mutations (9 novel) to the list of MLL2 mutations. Including patients described in our study, 21 splice-site mutations are now described in patients with KS (Supplementary Table 2). One patient in our study has a deletion spanning from the end of exon 49 to 10 bases into the intron, resulting in deletion of a single amino acid and abolishment of a splice donor site. Out of 19 MLL2 splice-site mutations, 7 have been shown to be de novo. Out of 10 splice variants, 2 are of uncertain pathogenicity. Resultant proteins of none of the MLL2 splice-site mutations have been characterised as yet. There are no obvious phenotypic differences between patients with splice-site and other mutations. Notably, one patient with c.13999+1G developed low-grade myofibrosarcoma and has been reported earlier.13
Nontruncating mutations
A total of 28 missense or in-frame insertions or deletions in MLL2, including 9 in this study, have been reported in 37 patients with KS. In our study, only those nontruncating changes that were proven to be de novo in at least one patient were considered to be definitely pathogenic. Using the same criteria in all the reported mutations, we can be certain about deleterious effect of 13 mutations in 22 patients (Supplementary Table 3). In 15 patients, 15 changes have not been checked for inheritance. It is therefore not possible to be certain about their pathogenicity.
Out of these 28 changes, one is a duplication of two amino acids resulting from a six base pair duplication in exon 48.7 p.I5497del, a single amino-acid deletion within the SET domain resulting from a mutation in exon 53, has been described in three patients, including one in this study. In our patient, this change was shown to be de novo and hence certainly pathogenic. Other 26/28 changes are missense.
Most of the pathogenic missense changes are concentrated toward the −COOH terminal of the protein. Out of the 37 (43.2%) patients with non-truncating non-splice-site mutations, 16 have changes in exon 48 (that codes for <7% of MLL2 protein). Five missense mutations in exon 48 have been described in more than one patient. This includes p.R5048C mutation that we found in a previously reported mother–daughter pair.14 Of note, exon 48 codes for LXXL motifs, one PHD domain and part of the FYRN domain.
We have also compiled a list of changes that are not known polymorphisms but have been seen concurrently in patients who have another definite pathogenic mutation (Supplementary Table 4). These are unlikely to be pathogenic but may be useful in understanding the role of various MLL2 residues. In this list we have also included variants that have been shown to be inherited from a normal parent. In our opinion, in the absence of stronger evidence to suggest variable penetrance in KS, such variants should be provisionally considered as benign. This degree of caution is especially important in clinical practice. Of note, four out of the five variants that we found concurrently with another definitely pathogenic mutation in this study are predicted to be highly damaging by AlignGVD and SIFT. This shows that in silico predictions for MLL2 are currently not entirely reliable, which is probably because the structure of MLL2 has not yet been realised. Hence, in a patient with KS, it is important to establish that a missense MLL2 change has arisen de novo before ascribing definite pathogenicity.
Facial KS morphology scores
Our group had contributed 18 patients to the study reported by Hannibal et al8 and, interestingly, MLL2 mutations were found in 17/18 patients (94.4%). Notably, these 18 patients were selected for having clinical features that are considered to be most typical of KS. The characteristics of mutations found in this group did not differ in any way from the larger cohort. Most previous studies have proposed that KS is genetically heterogeneous, but our experience indicates that most typical cases of KS are indeed due to MLL2 mutations. Furthermore, no strong genotype–phenotype correlation has emerged in previous studies comparing MLL2 mutation-positive and -negative patients. We hypothesised that facial KS morphology might be the most important difference between the two groups. We undertook a systematic approach to score facial KS morphology to test the genetic heterogeneity of ‘true’ KS.
Results of our facial KS morphology scores show that nearly all patients with ‘typical Kabuki face’ have mutations in MLL2 (Figures 2a, b and Table 1). Furthermore, there was no apparent trend correlating facial KS morphology scores with the type of mutation seen. Only 1 out of 15 MLL2 mutation-negative KS patients (KS110) scored high on the facial KS morphology score (93/115, mean 4.04; Figure 2d). Clinical history of this patient was also reminiscent of typical KS. Further work is needed to identify the underlying molecular mechanism in this child. Overall, this shows that based on facial dysmorphism, genetic heterogeneity of KS is not likely to be as large as previously expected.

Facial features of patients in this study. (a) Facial photograph of KS49, who has a MLL2 mutation, showing typical facial features of KS with interrupted high-arched eyebrows, long palpebral fissures, eversion of lateral part of lower eyelids, broad depressed nasal tip, large prominent earlobes and pillowed lower lip. (b) Facial photograph of KS45, who has a MLL2 mutation, showing deficient lateral eyebrows, long palpebral fissures, broad depressed nasal tip, large prominent earlobes, pillowed lower lip and oligodontia. (c) Facial photograph of KS36, who has a MLL2 mutation but is facially not typical for Kabuki syndrome. This demonstrates phenotypic variability of this condition indicating that MLL2 testing may be considered even in atypical patients. (d) Facial phenotype of KS110, which is typical of KS but MLL2 mutation was not found. She also has a history of severe feeding difficulties in infancy, coarctation of aorta, hypoplastic left ventricle, ventricular septal defect, aortic and sub-aortic stenosis, bilateral mild hydronephrosis, deep labial adhesions, right iris coloboma, bilateral choroido-retinal colobomas, single duplicated tooth, hypotonia and mild developmental delay.
A few patients who had relatively low facial KS morphology scores are not absolutely facially typical for KS but still have MLL2 mutations, demonstrating variability of facial phenotype in KS (see KS 62 and 36 in Table 1; Figure 2c). This indicates that MLL2 testing should be considered even in atypical KS patients.
In our and other geneticists' experience, it can be difficult to recognise KS in early infancy and in adulthood. Interestingly, for four patients whose two photographs taken at different ages were included in the slide show (Figures 3a–d), the scores for early childhood photographs corresponded better to the mutation status than those taken in infancy or adulthood.

Facial phenotype of KS changes with age. Both patients have MLL2 mutation. Mean and median scores for facial KS morphology are given in brackets. (a, b) Facial features of the same patient in neonatal period (2.64; 2) and later in infancy (4.45; 5, KS55). This patient has ‘grown into’ KS. (c, d) Facial features of the same patient in childhood (4.32; 4, KS85) and later in adulthood (3.23; 3). This patient has ‘grown out of’ KS.
Overall, the relationship between facial dysmorphism and MLL2 mutation status seems compelling, but these results should be interpreted with caution because by nature, such studies are subjective and open to many biases.
Genotype–phenotype correlation
Our finding of significant differences between feeding difficulties, early breast bud development or gynaecomastia, joint dislocations and cleft palate adds further to help delineate features that may increase the chances of finding MLL2 mutation in a patient (Table 2). Similar to two previous studies,6, 8 we also observed significant differences in rates of urogenital malformations. The range of feeding difficulties in MLL2 mutation-positive patients varied from just slow feeding or gastro-oesophageal reflux to needing nasogastric tube feeding and/or even gastrostomy. Most frequent joint dislocations were reported to involve the patella, hips, shoulders and elbows. One patient had bilateral thumb subluxations. A variety of palatal malformations like bifid or absent uvula, velopharyngeal insufficiency and central or bilateral cleft palate were seen in MLL2 mutation-positive patients.
MLL2 mutation-negative patients
Deep intronic mutations, large intragenic deletions or duplications or changes affecting the regulatory elements may not be recognised by sequencing of exons and exon–intron boundaries. Perhaps, MLPA or mRNA sequencing should be considered in typical MLL2 mutation-negative KS patients. However, from the facial KS morphology scores and emerging genotype phenotype correlation, it seems likely that many suspected KS patients without MLL2 mutation are likely to have unrelated conditions that may not be due to defects even in the same pathway. This is further indicated by the failure to find mutations in any genes associated with ASCOM complex in MLL2 mutation-negative patients in two other studies.6, 8
Four MLL2 mutation-negative patients with proposed KS in the study reported by Hannibal et al8 were found to have an alternative diagnosis by array comparative genomic hybridisation (a-CGH). One patient had a deletion on 5q encompassing NSD1, the second had a novel 19q23 deletion involving 20 genes, the third had a complex translocation involving chromosomes 8 and 18 and the fourth patient had mosaic trisomy 12. In our cohort we have not systematically performed a-CGH on all MLL2 mutation-negative patients but have found a deletion of 6q13, inherited from her affected mother, in one patient who we have always regarded to have atypical KS. Her mother also has similar clinical features. Similar deletions have been described in the literature.15 We therefore suggest a-CGH should be considered in MLL2 mutation-negative KS patients, if not done already.
Conclusions
Our work expands the known mutation spectrum of MLL2 mutations associated with KS. We have shown that the majority of mutations are truncating and the pathogenic missense mutations are commonly located in exon 48. We suggest that caution should be practised in assigning pathogenicity to missense MLL2 variants. Our compilation of mutation data available from the published literature may help laboratories offering MLL2 testing to ascribe significance to novel findings. Our mutation detection rate of ∼60% is a closer reflection of expected rate in clinical setting. Overall, this may provide insight into the structure and function of MLL2.
We show that KS can be phenotypically variable and therefore MLL2 testing should be considered even in atypical KS patients. Furthermore, we have demonstrated that MLL2 mutation-positive and -negative patients differ significantly in their facial dysmorphism and some clinical features. The majority of typical KS patients have MLL2 mutations, implying that the genetic heterogeneity of KS may be minimal. This work will also help in further studies to identify the genetic basis of patients with MLL2 mutation-negative KS.
References
Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I : A new malformation syndrome of long palpebralfissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr 1981; 99: 570–573.
Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T : Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr 1981; 99: 565–569.
Adam MP, Hudgins L : Kabuki syndrome: a review. Clin Genet 2005; 67: 209–219.
Ng SB, Buckingham KJ, Hannibal MC et al: Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 2010; 42: 790–793.
Paulussen ADC, Stegmann APA, Blok MJ et al: MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum Mutat 2011; 32: E2018–E2025.
Li Y, Bögershausen N, Alanay Y et al: A mutation screen in patients with Kabuki syndrome. Hum Genet 2011; 130: 715–724.
Micale L, Augello B, Fusco C et al: Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J Rare Dis 2011; 6: 38.
Hannibal MC, Buckingham KJ, Ng SB et al: Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet 2011; 155A: 1511–1516.
Dillon S, Zhang X, Trievel R, Cheng X : The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol 2005; 6: 227.
Ansari KI, Mandal SS : Mixed lineage leukemia: roles in gene expression, hormone signaling and mRNA processing. FEBS J 2010; 277: 1790–1804.
Goo Y-H, Sohn YC, Kim D-H et al: Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol Cell Biol 2003; 23: 140–149.
Mo R, Rao SM, Zhu Y-J : Identification of the MLL2 complex as a coactivator for estrogen receptor α. J Biol Chem 2006; 281: 15714–15720.
Shahdadpuri R, O'Meara A, O'Sullivan M, Reardon W : Low-grade fibromyxoid sarcoma: yet another malignancy associated with Kabuki syndrome. Clin Dysmorphol 2008; 17: 199–202.
Pottinger CC, Stiff RE, Holroyd JM, Davies SJ : Further evidence of dominant inheritance of Kabuki syndrome. Clin Dysmorphol 2009; 18: 215–217.
Van Esch H, Rosser EM, Janssens S et al: Developmental delay and connective tissue disorder in four patients sharing a common microdeletion at 6q13-14. J Med Genet 2010; 47: 717–720.
Acknowledgements
We are grateful to Mr and Mrs P Scales, their family and friends for funding and support received for this project through the Central Manchester University Hospitals NHS Foundation Trust, Kabuki Research Fund no. 629396. We also acknowledge the support of Manchester Biomedical Research Centre. We would like to show our appreciation to Dr Michael J Bamshad and Dr Katie J Buckingham from Seattle for providing us information for the primers for MLL2 sequencing. We thank all our patients, families and their clinicians for help in the research. We are grateful to following participants for scoring the facial KS morphology of patients at the M62 dysmorphology meeting: Dr Miranda Splitt, Dr Brian Wilson and Dr Gaik Siew (Newcastle upon Tyne); Dr Alan Fryer, Dr Lynn Greenhalgh and Dr Elizabeth Sweeney (Liverpool); Dr Angus Dobie, Dr Claire Searle, Dr Alison Krauss and Dr Jenny Thomson (Leeds); Dr Diana Johnson and Dr Meena Balasubramanian (Sheffield); and Dr Elizabeth Jones and Dr Sofia Douzgou (Manchester).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Banka, S., Veeramachaneni, R., Reardon, W. et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet 20, 381–388 (2012). https://doi.org/10.1038/ejhg.2011.220
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.220
Keywords
This article is cited by
-
Parallel functional annotation of cancer-associated missense mutations in histone methyltransferases
Scientific Reports (2022)
-
Identification of unique DNA methylation sites in Kabuki syndrome using whole genome bisulfite sequencing and targeted hybridization capture followed by enzymatic methylation sequencing
Journal of Human Genetics (2022)
-
Clinical heterogeneity of Kabuki syndrome in a cohort of Italian patients and review of the literature
European Journal of Pediatrics (2022)
-
Characteristics and surgical outcomes of cleft palate in kabuki syndrome: a case series of 11 patients
BMC Pediatrics (2021)
-
Kidney and urinary tract findings among patients with Kabuki (make-up) syndrome
Pediatric Nephrology (2021)
