
Abstract
Advances in genotyping technologies have contributed to a better understanding of human population genetic structure and improved the analysis of association studies. To analyze patterns of human genetic variation in Brazil, we used SNP data from 1129 individuals – 138 from the urban population of Sao Paulo, Brazil, and 991 from 11 populations of the HapMap Project. Principal components analysis was performed on the SNPs common to these populations, to identify the composition and the number of SNPs needed to capture the genetic variation of them. Both admixture and local ancestry inference were performed in individuals of the Brazilian sample. Individuals from the Brazilian sample fell between Europeans, Mexicans, and Africans. Brazilians are suggested to have the highest internal genetic variation of sampled populations. Our results indicate, as expected, that the Brazilian sample analyzed descend from Amerindians, African, and/or European ancestors, but intermarriage between individuals of different ethnic origin had an important role in generating the broad genetic variation observed in the present-day population. The data support the notion that the Brazilian population, due to its high degree of admixture, can provide a valuable resource for strategies aiming at using admixture as a tool for mapping complex traits in humans.
Similar content being viewed by others


Introduction
The advances in genotyping technologies have provided important and considerable insights regarding our views of human population structure. The knowledge of patterns of genetic variation within and among human populations have contributed to a better understanding of the relationship between genetics and ethnicity, as well as improved the design and analysis of case–control association studies. Although there are several studies that have investigated the genetic structure of non-Caucasian populations, including individuals of African, African Americans, Asian, and Native American ancestry, most studies have primarily focused on individuals of European ancestry.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 Therefore, coverage of the global human population remains incomplete with populations from South America being underrepresented in the databases of human genetic variation. Included in these understudied populations are individuals from Brazil, a country of almost 200 million people, which represents approximately 52% of the South American population and 3% of the world's population.
Historically, the Brazilian population always experienced large degrees of intermarriage between ethnic groups, and Brazilians are known to be heavily admixed with Amerindian, European, and African ancestries. In general, Brazilians trace their origins to the original Amerindians and two main sources of immigration: Africans and Europeans.13, 14 In the five geographical regions of Brazil (North, Northeast, Center–West, Southeast, and South), Northern Brazilians are mostly of Amerindian ancestry, with some African ancestry. Current inhabitants of Northeast and Center–West are mostly of African origin, although some individuals whose ancestors migrated from Southern Brazil can trace their roots to Europe. Southern and Southeastern Brazilians are mostly of European origin. However, individuals of African and Asian descent are also found in several localities of the Southeast. For decades, new immigrants, as well as migrants from other parts of Brazil, have flocked to Southeast Brazil where intermarriage between individuals of different ancestry is very common. The goals of the present work are to: (i) identify patterns of population structure among the Southeast Brazilian population enabling individuals from this region to be included in future studies of genetic variation, (ii) to identify marker panels that can effectively capture the variation revealed by dense genotyping from samples of the Southeast Brazilian population and samples from the 11 populations of the HapMap Project, Phase III, which include individuals of Asian, African, European, and Mexican ancestry, and (iii) assess global and local ancestry inferences of the Southeast Brazilian population.
Materials and methods
Datasets and preprocessing steps
Analysis was performed considering samples of the Southeast Brazilian population (BRZ), as well as samples from the following 11 populations of the HapMap database, Phase III: African ancestry in Southwest (ASW), Utah residents with Northern and Western European ancestry from the CEPH collection (CEU), Han Chinese in Beijing, China (CHB), Chinese in Metropolitan Denver, Colorado (CHD), Gujarati Indians in Houston, Texas from the western state of Gujarat in India (South Asia) (GIH), Japanese in Tokyo, Japan (JPT), Luhya in Webuye, Kenya (LWK), Mexican ancestry in Los Angeles, California (MEX), Masai in Kinyawa, Kenya (MKK), Tuscans in Italy (TSI), and Yoruba in Ibadan, Nigeria (YRI). International HapMap Project, Phase III is available at http://www.sanger.ac.uk/humgen/hapmap3.
The Southeast Brazilian population samples are from a study conducted with trios of individuals (mother, father, and son or daughter), whose children have a congenital heart disease and parents do not. All individuals are from the general urban population of Sao Paulo, the largest metropolitan area of the country. In the present analysis, we have only used data from those unrelated individuals (mothers and fathers). These individuals were enrolled in the current study at the Heart Institute of the University of Sao Paulo. Genotyping for these samples was performed using the Affymetrix SNP array 6.0 platform (Affymetrix, Santa Clara, CA, USA). All subjects gave verbal and written consent. The present protocol was approved by the University of Sao Paulo Medical School IRB (CAPPesq). Samples from the HapMap were genotyped using two platforms, Affymetrix SNP 6.0 and Illumina Human 1M arrays (Illumina, San Diego, CA, USA). More details from the HapMap populations are available from the HapMap Project webpage. Only unrelated individuals were considered in the present analysis. Only SNPs located on the autosomal chromosomes and successfully genotyped in all populations were used for this analysis.
SNPs that were not accurately assessed on the Affymetrix 6.0 array were excluded from the final analysis. That is, we removed, separately for each of the 12 populations, SNPs with more than 5% missing genotype, SNPs that were not in Hardy–Weinberg equilibrium (P≤10−4), and also those with a minor allele frequency less than or equal to 0.01. At the end of these steps, 365 116 autosomal SNPs, shared by all 12 population data sets and 1129 unrelated individuals representing the 11 HapMap populations (n=991) and the Brazilian population (n=138), remained.
Statistical analysis
We used Principal Components Analysis (PCA), a dimensionality reduction technique,1, 2 to analyze the data. For each population k, the data set consists of nk unrelated subjects, where each subject has m biallelic SNPs common for all populations. Data for all 12 populations were then displayed in a matrix G of dimension m by n with n = ∑k = 112 nk. The values 0, 1, 2, or empty, correspond to the genotypic information assigned to each SNP.2 After mean-centering and normalizing each row i of the matrix G, n eigenvalues and n corresponding eigenvectors (axes of variation) were calculated, using the covariance matrix of individuals ψ=G′G. Plots of the eigenvectors associated with the largest eigenvalues were then used to investigate the structure of the populations under analysis. PCA was run without the removal of outliers and without eliminating SNPs in linkage disequilibrium.
To investigate whether a smaller number of SNPs could effectively capture the variation revealed by the 365 116 common SNPs, we built three panels of markers. The first panel has 250 SNPs, consisting of the top 50 SNPs retained from each of the top five axes of variation. SNPs were ranked on the basis of their loading scores (in absolute value) obtained from the axes of variation. The second and third panels were obtained by retaining the top-ranked 100 and 150 SNPs from each of the same top five axes, respectively. As there were no common SNPs among those retained, the total number of SNPs left in each panel was 250, 500, and 750, respectively. The relationship between the different populations was also investigated by calculating the Fst statistic, a metric representation of the effect of population subdivision15, 16 for each pair of populations, using the SNPs in the three panels, and also the 365 116 common SNPs. Fst statistic is often expressed as the proportion of genetic diversity due to allele frequency differences among populations. A zero value implies that the two populations are interbreeding freely and a value of one that the two populations are completely separate.
For global ancestry analysis, we applied the model-based STRUCTURE program17 to estimate the admixture proportion for the BRZ samples. This was done by applying the STRUCTURE program to two different pooled data sets consisting of four reference populations each (CEU, YRI, MEX, and BRZ, for model 1) and (TSI, ASW, MEX, and BRZ, for model 2), without informing the program which samples were the reference samples. The reason for selecting model 2 was based on the smallest Fst values obtained between the BRZ and HapMap, Phase III samples of Caucasian and African origin. As seen in Figure 3, the performance of the first two PCs in each of the two different pooled data sets is similar. We allowed the program in such an unsupervised mode to infer the underlying ancestral populations, as well as the ancestral proportion for each subject. The number of ancestral populations K was fixed at 3, 4, 5, and 7. For a given K, we ran STRUCTURE 10 times with different random seeds (10 000 iterations for burn-in phase, and 10 000 iterations for Markov chain optimization and recorded L(K), the log likelihood of the data given K, from each run. We used the metric ΔK to find the optimal K, which is selected to have the largest ΔK value.18 The inferred number of ancestral populations for the pooled data was 3.
Analyses described above were carried out using the publicly available STRUCTURE,17 and EIGENSTRAT2, 7 software packages.
Results
Principal components analysis
PCA using the 12 populations showed pronounced patterns of genetic variation within and amongst the populations. To visualize these patterns graphically, we shall consider the top three axes of variation chosen on the basis of their eigenvalues (Figure 1).

Eigenvalues associated with the 20 first PCs (axes of variation) obtained from the PCA, in which all common SNPs were used.
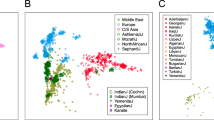
The two and three most informative axes of variation, PC1 and PC2 (Figure 2a), and PC1, PC2, and PC3 (Figure 2b), can resolve the 11 populations available in the HapMap study. That is, despite some overlap, we observed that the individuals from the 11 HapMap populations were clearly separated by their different ancestries of origin (African, Asian, European, and Mexican). Asian populations were tightly clustered and distinct from the African and European populations. The Southeast Brazilian population formed a continuum between Europeans and Africans, with some overlap of the Mexican population. The continuum of genotypes observed in the Brazilian population is consistent with the high degree of intermarriage between individuals of the European and African descent.

Projection of 1129 individuals from 11 populations of the HapMap Project, Phase III, and the Brazilian population on their (a) first and second, and (b) first, second, and third axes of variation obtained from PCA, which used 365 116 SNPs. ASW, African ancestry in Southwest; CEU, Utah residents with Northern and Western European ancestry from the CEPH collection; CHB, Han Chinese in Beijing; China, CHD, Chinese in Metropolitan Denver, Colorado; GIH, Gujarati Indians in Houston, Texas; JPT, Japanese in Tokyo, Japan; LWK, Luhya in Webuye, Kenya; MEX, Mexican ancestry in Los Angeles, California; MKK, Masai in Kinyawa, Kenya; TSI, Tuscans in Italy; YRI, Yoruba in Ibadan, Nigeria; and BRZ, Brazilians in São Paulo, Brazil.
Fst statistic results
The Fst statistic was calculated for all population pairs using the 365 116 common SNPs (Table 1). Small Fst values (0.001 to 0.008) were found for each pair of Asian populations (CHB, CHD, and JPT), indicating less pronounced genetic differences between these populations. Similarly, each pair of African populations (ASW, LWK, MKK, and YRI) is separated by low Fst scores. Greater Fst distances (0.128 to 0.168) were observed between Asian and African populations. Populations with European ancestry (CEU and TSI) are also separated by small Fst values (0.003). Three distinct clusters of ancestral populations (Asian, African, and European) are distinguished by Fst scores. MEX and GIH populations are closer to the European cluster than to the African cluster as measured by Fst distance. Fst scores confirm that the Southeast Brazilian population is close to both the European, African, and Mexican populations.
Ancestry informative markers
Small sets of ancestry informative markers (AIMs) that can provide substantial substructure information have been the focus of several studies.19, 20, 21 AIM sets consisting of 200 markers or less can map ancestral origin to Africa, Europe, or Asia. We considered three panels of markers. SNPs on each panel were selected on the basis of their loading scores obtained from a PCA performed on the covariance matrix of the SNPs. The first panel has 250 SNPs consisting of 50 SNPs with highest loading scores (in absolute value) on the top five axes of variation. The second and third panels retained 100 and 150 SNPs, respectively, of the top five axes of variation, and have 500 and 750 markers, respectively. Plots of the two first axes of variation (PC1 and PC2) were obtained by performing PCA for each of the three panels of SNPs (data not shown). The 250 SNP set reproduced the stratification observed with the entire 365 116 SNP set (Figure 2). The 500 and 750 SNP set produced results that were indistinguishable from the 250 SNP set. The chromosomal distribution of the 500 SNP set was uniform. Although the magnitude of the Fst values varied, the same pattern could be observed for all three panels of markers (Table 2). All three SNP marker panels captured the variation revealed by the entire >300 000 SNP set. Indeed, calculation of the pairwise Spearman correlation coefficient between the four Fst matrices yielded results always higher than 0.964.
Global ancestry inference of the Brazilian population
Global ancestry inference of the studied samples was able to determine mean ancestries for Amerindian, African, and European. For such, we have first recalculated Eigenstrat principal components, using two different subsets of HapMap samples as ‘ancestral’ populations. In the first model, we have used the CEU, YRI, and MEX samples to represent, respectively, a Caucasian, African, and Amerindian ancestral population. In the second model, we used the TSI, ASW, and MEX samples to represent such populations. The reason for using the first model was because of the common use of these as ancestral populations in most of the earlier reports. In the second model, we have used the populations with smallest Fst pairwise differences with the BRZ sample. No significant differences between these two models were observed (Figure 3). Structural analysis, using the 100 most important SNPs from PC1 and PC2, from these two models is presented in Figure 4. In our sampled individuals from the Brazilian Southeast region, mean values were 0.15, 0.24, and 0.61, respectively, for Amerindian, African, and European ancestries for Model I markers, and 0.17, 0.27, and 0.56, respectively, for Amerindian, African, and European ancestries for Model II markers (Figure 4).

Projection of individuals from three potentially ancestral populations of the HapMap Project, Phase III, and the Brazilian population on their first and second axes of variation (PCs) using Model 1=YRI, CEU, MEX, and BRZ, and Model 2=ASW, TSI, MEX, and BRZ.

Proportion of membership of each pre-defined population in each of the three clusters. (a) Triangular plot of the genomic proportions of African, European, and American ancestry, of the sampled populations from Model I (CEU, YRI, and MEX). (b) Barplot structure analyses with admixture model for sampled populations from Model I. (c) Triangular plot of the genomic proportions of African, European, and American ancestry, of the sampled populations from Model II (TSI, ASW, and MEX). (d) Barplot Structure analyses with admixture model for sampled populations from Model II. Red, blue, and green, represent the proportions of inferred ancestry from European, African, and American ancestral populations. (MEX, Mexican ancestry in Los Angeles, California; CEU, Utah residents with Northern and Western European ancestry from the CEPH collection; YRI, Yoruba in Ibadan, Nigeria; TSI, Tuscans in Italy; ASW, African ancestry in Southwest; and BRZ, Brazilians in São Paulo, Brazil).
Discussion
We have compared the genotypic variation of 365 116 SNPs among 1129 unrelated individuals of five continents (Asia, Europe, Africa, and North and South America) to individuals from Southeast Brazil. We demonstrate that this population is a highly admixed population and quite distinct from other HapMap populations. Principle component analyses demonstrate extensive of intermarriage between individuals of African and European descent. This intermarriage occurred between 1500 and the present day reflecting about 20 generations of intermarriage. Thus, the genomes of Brazilian individuals consist of chromosomal segments of distinct ancestry with substantial European and African-related admixture. These findings will have important implications for the correct design and analytical planning of studies exploring complex traits in this population. We expect that the large degree of admixture observed in the Southeast Brazilian population can be exploited for the gene mapping of important disease loci.
The study cohort was collected in Southeast Brazil, in Sao Paulo state. Individuals of African, Amerindian, and perhaps Asian ancestries, may be underrepresented in this study, as individuals with European ancestry comprise a majority in this region. Thus, additional analyses using larger and random samples that can cover all five Brazilian regions might perhaps show an even more pronounced degree of genetic variation than the one suggested by our analysis. Whether the same degree of intermarriage will be observed in other parts of Brazil or other parts of Latin America will be addressed in future studies.
New dense genotyping data from other forthcoming Brazilian studies will determine whether the same pattern of extensive genetic admixture exists in other parts of Brazil.
References
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D : Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
Seldin MF, Shigeta R, Villoslada P et al: European population substructure: clustering of northern and southern populations. PLoS Genet 2006; 2: e143.
Paschou P, Ziv E, Burchard EG et al: PCA-correlated SNPs for structure identification in worldwide human populations. PLoS Genet 2007; 3: 1672–1686.
Heath SC, Gut IG, Brennan P et al: Investigation of the fine structure of European populations with applications to disease association studies. Eur J Hum Genet 2008; 16: 1413–1429.
Paschou P, Drineas P, Lewis J et al: Tracing sub-structure in the European American population with PCA-informative markers. PLoS Genet 2008; 4: e1000114.
Price AL, Butler J, Patterson N et al: Discerning the ancestry of European Americans in genetic association studies. PLoS Genet 2008; 4: e236.
Biswas S, Scheinfeldt LB, Akey JM : Genome-wide insights into the patterns and determinants of fine-scale population structure in humans. Am J Hum Genet 2009; 84: 641–650.
Xing J, Watkins WS, Witherspoon DJ et al: Fine-scaled human genetic structure revealed by SNP microarrays. Genome Res 2009; 19: 815–825.
McEvoy BP, Montgomery GW, McRae AF et al: Geographical structure and differential natural selection among North European populations. Genome Res 2009; 19: 804–814.
Auton A, Bryc K, Boyko AR et al: Global distribution of genomic diversity underscores rich complex history of continental human populations. Genome Res 2009; 19: 795–803.
Adeyemo A, Gerry N, Chen G et al: A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet 2009; 5: e1000564.
Goncalves VF, Carvalho CM, Bortolini MC, Bydlowski SP, Pena SD : The phylogeography of African Brazilians. Hum Hered 2008; 65: 23–32.
Suarez-Kurtz G : Pharmacogenomics in Admixed Populations. Landes Bioscience: Austin, 2007.
Wright S : Genetical structure of populations. Nature 1950; 166: 247–249.
Duan S, Zhang W, Cox NJ, Dolan ME : FstSNP-HapMap3: a database of SNPs with high population differentiation for HapMap3. Bioinformation 2008; 3: 139–141.
Falush D, Stephens M, Pritchard JK : Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 2003; 164: 1567–1587.
Wang Z, Hildesheim A, Wang SS et al: Genetic admixture and population substructure in Guanacaste Costa Rica. PLoS One 2010; 5: e13336.
Yang N, Li H, Criswell LA et al: Examination of ancestry and ethnic affiliation using highly informative diallelic DNA markers: application to diverse and admixed populations and implications for clinical epidemiology and forensic medicine. Hum Genet 2005; 118: 382–392.
Kosoy R, Nassir R, Tian C et al: Ancestry informative marker sets for determining continental origin and admixture proportions in common populations in America. Hum Mutat 2009; 30: 69–78.
Enoch MA, Shen PH, Xu K, Hodgkinson C, Goldman D : Using ancestry-informative markers to define populations and detect population stratification. J Psychopharmacol 2006; 20: 19–26.
Acknowledgements
We thank the CNPq (Brazil, Grant 150653/2008–5) for partial financial support (SRG). This work was supported by FAPESP (Grant 2007/58150-7), and Hospital Samaritano, Sao Paulo.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Giolo, S., Soler, J., Greenway, S. et al. Brazilian urban population genetic structure reveals a high degree of admixture. Eur J Hum Genet 20, 111–116 (2012). https://doi.org/10.1038/ejhg.2011.144
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.144
Keywords
This article is cited by
-
Black and non-black population: investigation of the difference in butyrylcholinesterase activity in a healthy population in Salvador, Bahia
Irish Journal of Medical Science (1971 -) (2023)
-
Somatic targeted mutation profiling of colorectal cancer precursor lesions
BMC Medical Genomics (2022)
-
Association of Toll-like receptors polymorphisms with the risk of acute lymphoblastic leukemia in the Brazilian Amazon
Scientific Reports (2022)
-
Genetic ancestry inferred from autosomal and Y chromosome markers and HLA genotypes in Type 1 Diabetes from an admixed Brazilian population
Scientific Reports (2021)
-
Association between vitamin D plasma concentrations and VDR gene variants and the risk of premature birth
BMC Pregnancy and Childbirth (2020)
