
Abstract
Among inherited retinal dystrophies, autosomal recessive retinitis pigmentosa (arRP) is the most genetically heterogenous condition with 32 genes currently known that account for ∼60 % of patients. Molecular diagnosis thus requires the tedious systematic sequencing of 506 exons. To rapidly identify the causative mutations, we devised a strategy that combines gene mapping and phenotype assessment in small non-consanguineous families. Two unrelated sibships with arRP had whole-genome scan using SNP microchips. Chromosomal regions were selected by calculating a score based on SNP coverage and genotype identity of affected patients. Candidate genes from the regions with the highest scores were then selected based on phenotype concordance of affected patients with previously described phenotype for each candidate gene. For families RP127 and RP1459, 33 and 40 chromosomal regions showed possible linkage, respectively. By comparing the scores with the phenotypes, we ended with one best candidate gene for each family, namely tubby-like protein 1 (TULP1) and C2ORF71 for RP127 and RP1459, respectively. We found that RP127 patients were compound heterozygous for two novel TULP1 mutations, p.Arg311Gln and p.Arg342Gln, and that RP1459 patients were compound heterozygous for two novel C2ORF71 mutations, p.Leu777PhefsX34 and p.Leu777AsnfsX28. Phenotype assessment showed that TULP1 patients had severe early onset arRP and that C2ORF71 patients had a cone rod dystrophy type of arRP. Only two affected individuals in each sibship were sufficient to lead to mutation identification by screening the best candidate gene selected by a combination of gene mapping and phenotype characterization.
Similar content being viewed by others


Introduction
Photoreceptor degenerations are the leading cause of inherited blindness.1 This is partly explained by the extreme genetic heterogeneity of these conditions with 160 genes currently registered in the RetNet database (www.sph.uth.tmc.edu/retnet), reflecting the vast repertoire of genes necessary for photoreceptor function. Concurrently, there is a variety of phenotypes caused by photoreceptor loss, which are classified in many clinical entities depending on the presence or absence of systemic involvement, the severity and course of the disease, the location and shape of retinal lesions and deposits, the involvement of either rod, cone or both photoreceptors, and the type of electrical responses to light stimuli. The most frequent clinical entity, non-syndromic retinitis pigmentosa (RP), is also the most genetically heterogeneous, with 51 disease causing genes being currently known in this condition. From these, 32 cause autosomal recessive (ar) forms of the disease, accounting for 50 to 60 % of all arRP cases.1
Molecular diagnosis in arRP thus requires the systematic sequencing of 506 exons to cover the 32 genes. This is a tremendous task with conventional sequencing methods, which in addition can miss mutations located in non-coding regions. Exome sequencing using high throughput sequencing technologies are powerful new methods, but they still remain costly. Alternatively, homozygosity mapping in inbred, multiplex families or isolated cases, is time saving by readily pointing at only one or a few regions containing an already known disease causing gene or at new genes/loci.2 This strategy has also been successful in a variable proportion of cases from outbred families, which carry a homozygous mutation, because of inbreeding encountered in some populations.2, 3, 4 However, homozygous regions unlinked to disease phenotypes are common in the human genome,5 and therefore may erroneously suggest false candidate regions. In addition, the majority of families originating from countries with mixed populations are outbred and the affected patients carry compound heterozygous mutations. For these families, homozygosity mapping will thus remain uninformative.
Here we devised a strategy based on gene mapping in non-consanguineous families to search for mutations in known genes. In two families, we performed a genome-wide SNP analysis and found many candidate chromosomal regions. By determination of a score from genotyping results and by assessment of phenotype features, we selected one candidate gene in each family. This process allowed to identify the novel mutations in TULP1 and in the recently described C2ORF71, thus evidencing the potential interest of this method for mutation finding in small outbred families.
Methods
Patients and clinical investigations
Two non-consanguineous French families (RP127 and RP1459) with non-syndromic RP and evidence of autosomal recessive inheritance were recruited (Figures 1a and c). In RP127, two out of six siblings were affected; parents and offsprings from the two affected patients were normal. In RP1459, two out of three sisters were affected; parents were normal. Informed consent and blood samples were obtained from family members. The investigators followed the tenets of the Declaration of Helsinki.

Pedigrees and sequence analysis in two families segregating autosomal recessive retinitis pigmentosa. (a, c) Pedigree of families RP127 (a) and RP1459 (c); blackened symbols are affected individuals, mutation genotype for TULP1 (a) or C2ORF71 (c) is indicated under each family member (‘+’ means a wild-type allele). (b, d) Electropherograms; the normal sequence is written in black italic and the mutated nucleotides are in red. (b) TULP1 sequence for each of the two mutations in patient (indicated above) compared with wild type (normal) is shown. (d) C2ORF71 sequence for each of the two mutations in either father or mother (indicated above) is shown and compared to that of patient who carry both mutations in exon 1 and of wild-type individual (normal).
Patients had standard ophthalmologic examination (refractometry, visual acuity, slit-lamp examination, applanation tonometry and fundoscopy). Kinetic visual fields were determined with a Goldman perimeter with targets V4e, III4e and I4e. OCT measurement of the macula was performed using an OCT-3 system (Stratus model 3000, Carl Zeiss Meditec, Dublin, CA, USA) with the software version 3.0. Autofluorescence measurements were obtained with the HRA2 Heidelberg retinal confocal angiograph (Heidelberg Engineering, Dossenheim, Germany) and fundus pictures were taken. Full-fields ERG was recorded using a Ganzfeld apparatus (Metrovision, Pérenchies, France) with a bipolar contact lens electrode on maximally dilated pupils according to the ISCEV protocol.6
Genotyping and mapping
Genomic DNA was extracted from 10-ml peripheral blood samples by a standard salting out procedure.7 In all, 11 members of the RP127 family and the 3 sisters of the RP1459 family were genotyped for 262 270 SNPs (GeneChip Mapping 250K Nsp Array, Affymetrix, Santa Clara, CA, USA) at the Centre National de Génotypage (http://www.cng.fr), Evry, France or at DNAVision, Charleroi, Belgium. Results were analyzed using TASE (for transmitted allele search engine), a software designed in our laboratory (http://www.inmfrancetools.com/TASE) to search for common genotypes in all affected individuals (CGAA test). This test compares every SNP between each individual in the family, and assigns one of three possible states to each SNP: (i) excluding SNP (affected individuals have different genotypes), (ii) neutral SNP (all affected individuals share the same genotype with some non-affected individuals) and (iii) qualifying SNP (all affected individuals share the same genotype while non-affected individuals carry another genotype). Candidate chromosomal regions were then defined as stretches of neutral and/or qualifying SNPs encompassing regions that were ≥5 Mb and assigned centromeric and telomeric boundaries defined by two consecutive excluding SNPs. For each chromosome, the results were displayed as a chart showing the status of each SNP (see http://www.inmfrancetools.com/TASE), and the candidate regions were then compared with the position of known genes and loci for retinal inherited diseases according to the RetNet database (http://www.sph.uth.tmc.edu/retnet).
Selection of candidate genes
This was based on data issued from gene mapping and phenotype characterization. For a given candidate region, the probability to contain the causative gene usually increases with its coverage (average number of SNPs per megabase) and the qualifying rate (QR; percentage of qualifying SNPs over the total number of SNPs in the region). We calculated a qualifying score (QS) defined as the product of coverage × QR/10 that took into account these two parameters. Regions with the highest QSs were considered as those with the highest probability to contain the causative gene. In parallel, known retinal disease genes present in the candidate regions were listed, and the phenotypes usually caused by mutations in these genes were compared with the phenotype observed in the patients for each family. For each gene, the comparison was qualified as ‘concordant’ when the candidate genes were indeed responsible for an arRP form similar to that observed in the families or as ‘non-concordant’ when candidate genes were responsible for RP (or non-RP) phenotypes different from that observed in the family. Only a few occurrences of concordant phenotypes with the highest QSs remained allowing for the selection of a few candidate genes to sequence.
Mutation screening
All exons and exon–intron boundaries of TULP1 (GenBank accession #NM_003322.3) and C2ORF71 (GeneBank accession #NM_001029883.1) were sequenced. Primer pairs chosen for the 15 TULP1 exons and the 2 C2ORF71 exons are available on request. Each PCR was performed in a 25-μl reaction mixture containing 50 ng genomic DNA, 2 mM MgCl2, 200 μ M dNTPs, 0.2 μ M of each primers and 1 U of Taq DNA Polymerase AmpliTaq Gold (Applied Biosystems, Foster City, CA, USA) in its appropriate buffer. Following the first denaturation at 95 °C for 9 min, amplification was carried out for 35 cycles at 95 °C for 30 s, at the melting temperature (Tm) of the primers (56–60 °C) for 1 min and at 72 °C for 1 min, ending with a final extension step at 72 °C for 10 min. PCR products were purified with ExoSap-it clean up (Amersham Biosciences, Piscataway, NJ, USA) and sequenced using the BigDye Terminator cycle sequencing ready reaction kit V3.1 on an Applied Biosystems 3130xL genetic analyzer (both Applied Biosystems) following manufacturer's instructions. Sequencing results were analyzed by alignment with the Clustalw program (version 1.83).
Results
Clinical description
Family RP127
Proband (II.5) had night blindness and visual field defects since early childhood. At time of presentation (age 42), she had moderate myopia (−1.25(−1.50; 40°) OD; −1.00(−1.75; 10°) OS). She was counting fingers on the right eye and had hand motion on the left eye. She had posterior subcapsular cataract and intraocular pressure was normal at 14 mm Hg on both eyes. Her fundus showed a bilateral macular atrophy and dense bone spicule-shaped pigment deposits in retinal periphery (Figures 2a and b). Retinal vessels were highly attenuated and optic disks were pale. She was seen again at age 55 and had only light perception in both eyes. Visual field was undetectable.

Fundus images of affected patients in family RP127. (a) Right and (b) left eye posterior poles of patient II.5 at age 42 showing round shape atrophy of the macula, advanced atrophy of the peripheral retina, pigment deposits and narrowing of retinal vessels. (c) Superior and (d), inferior retina in the right eye of patient II.2 at age 54 showing major atrophy of the whole retina including retinal periphery and macula, with pigment deposits, tenuous retinal vessels and waxy pale optic disks.
Her affected eldest brother (II.2) also had night blindness and visual field defects since early childhood, leading to the diagnosis of RP at age 5. He could never drive. Yet, he was able to read until the age of 33, at which time he had cataract surgery but no improvement in visual acuity. At 40, he was virtually blind and needed a white stick to move outside. At time of presentation (age 54) he had no light perception in the right eye and faint light perception in the left eye. Intraocular pressure was normal at 14 mm Hg in both eyes. The macula had a yellowish, disorganized aspect and many bone spicule-shaped pigment deposits were present in mid periphery (Figures 2c and d). Retinal vessels were hardly visible and he had waxy optic disks. He died from repeated cardiac infarctions.
Mother (I.2), patients’ sibs (II.1, II.3, II.4, II.6) and patients’ children (III.1, III.2, III.3, III.4) were examined; no signs of RP were detected. We concluded that patients II.2 and II.5 had severe arRP.
Family RP1459
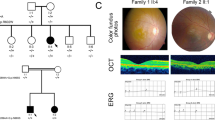
Proband (II.2) had moderate dyschromatopsia since early childhood. Later on, at age 14, she noticed moderate night blindness and photophobia. She had difficulties to distinguish fine objects in far sight but had no reading difficulties. She did not complain of loss in the peripheral visual field and was able to drive her car in daytime. At the time of presentation (age 25), her visual acuity was moderately decreased at 20/30 on both eyes with myopic refraction −6.75(−1.75; 45°) OD; −5.75(−0.75; 110°) OS. Intraocular pressure was normal at 14 mm Hg on both eyes. Lenses were transparent. Her fundus showed slightly discolored foveas with loss of foveal reflex, slightly pale optic disks and vascular attenuation (Figure 3a). There were no pigment deposits or retinal atrophy. Autofluorescence of macula was moderately heterogeneous whereas that of peripheral retina was normal (Figure 3b). OCT scans showed thinning of the retina with loss of the IS/OS line in both fovea and macula (Figure 3e). Goldman perimetry showed that peripheral isopter V4e was normal (90° temporal, 60° nasal) but there was a relative central scotoma on 20° around fixation at I4e in both eyes. Moderate tritanopia was confirmed on desaturated 15 HUE test. ERG testing showed barely detectable responses both in scotopic (responses only at maximal stimulation) and photopic conditions (responses only at 30-Hz flickers) (Figure 3g).

Clinical findings in family RP1459. (a, c) Fundus photographs and (b, d), fundus autofluorescence of left eye of patients II.2 (a, b) and II.1 (c, d) showing slight discoloration of fovea, loss of normal foveal reflex and moderate narrowing of retinal vessels (a, c) and alteration of the foveal retinal pigment epithelium (b, d). (e, f) OCT scans of left eye of patients II.2 (e) and II.1 (f) showing thinning of the retina with relative preservation of foveal photoreceptors. (g) Electroretinogram recordings showing the dramatic reduction of light-adapted responses of patients II.1 and II.2 with relatively conserved dark-adapted response of patient II.1, compared with normal responses in a control individual (normal) and to slightly decreased light-adapted responses in heterozygous carriers II.3, I.1 and I.2.
Affected sister (II.1) had moderate dyschromatopsia and night blindness since early childhood. At age 20 she noticed moderate photophobia. She had slight reading difficulties. She did not complain of loss in the peripheral visual field and was able to drive her car both in day and night-time. At the time of presentation (age 30), her visual acuity was moderately decreased at 20/40 on both eyes with +1.50(−1.75; 95°) OD; +1.50(−1.75; 70°) OS. Lenses were transparent. Her fundus showed slightly discolored foveas with loss of foveal reflex, normal optic disks and moderate vascular attenuation (Figure 3c). There were no pigment deposits or retinal atrophy. Autofluorescence of macula was moderately heterogeneous, whereas that of peripheral retina was normal (Figure 3d). OCT scans showed thinning of the retina with loss of the IS/OS line in the macula and relative sparing in the fovea (Figure 3f). Goldman perimetry showed that peripheral isopter V4e was moderately decreased (70° temporal, 50° nasal) with relative central scotoma on 20° around fixation at I4e in both eyes. There was non-systematized dyschromatopsia confirmed on desaturated 15 HUE test. ERG testing showed barely detectable responses in photopic conditions but responses in scotopic conditions were still present even for the lowest stimuli, although attenuated (Figure 3g).
The parents (II.1, II.2) and sister II.3 had no symptoms, normal visual acuity and funduscopy, indicating that they did not have RP. Yet, they all had slightly decreased photopic responses at ERG (Figure 3g). On the basis of dyschromatopsia, decreased visual acuity while peripheral visual field was normal, and ERG finding in II.1, we concluded that affected sisters II.1 and II.2 had the cone rod dystrophy form of arRP, with some intrafamilial variations.
Gene mapping, selection of candidate genes and mutation identification
Family RP127
Genome-wide SNP genotyping was performed on the 11 members of family RP127. Using the TASE software, we found that 33 chromosomal regions were common to the two affected patients, with an average length of 21 Mb (range: 5–68 Mb) and coverage of 75.5 SNPs/Mb (range: 0.6–133 SNPs/Mb; Table 1). The QR and QS were first calculated for the six members of the sibship (generation II). The five best QSs (range: 233.1–56.5) were found to correspond (from highest to lowest QS) to chromosomes 3, 18, 6, 11 and 5. Adding healthy members of the family from generations I and III should decrease the probability that affected sibs had common genotypes with another member of the family only by chance. When the mother I.2 (generation I) and the four members of generation III were added (11 members), the value of the QS therefore dropped but highlighted the genotype specific to the affected individuals. The order of the five best QSs (range 20.4–9.5) changed to the chromosome 11 region first (QS 20.4), followed by chromosome 6 (QS 14.1) and then 1, 3 and 5. We then listed all possible genes and phenotypes for each chromosomal region and compared them with the phenotype of affected members of family RP127. In all, 15 of the 33 regions contained 31 candidate genes previously reported in inherited retinal diseases (Table 1). When comparing phenotypes caused by these candidate genes with the severe non-syndromic arRP of the two affected family members, it was found that RPE65, ABCA4, SNRNP200, RHO, PROM1, TULP1, IMPDH1, RGR, SPATA7, TTC8, CNGB1 and CA4 were concordant. Of these, only TULP1 was in the chromosomal regions with the five best QS. As TULP1 was repeatedly reported in severe RP cases and was also the candidate gene of chromosome six region with the second highest QS, we sequenced all TULP1 coding exons and exon–intron boundaries in the 11 family members. We found two nucleotide changes, c.932G>A in exon 10 and c.1025G>A in exon 11, resulting in two novel amino acid substitutions, p.Arg311Gln and p.Arg342Gln, respectively (Figure 1b). These substitutions segregated with the RP phenotype (Figure 1a). Indeed, affected patients II.2 and II.5 carried both mutations in trans, whereas healthy individuals carried either only one mutation or wild-type alleles. These changes were not found in 57 unrelated controls, indicating that they were likely to be disease-causing mutations.
Family RP1459
To test whether this strategy would be efficient in a smaller sibship, we performed genome-wide SNP genotyping on the three sisters of family RP1459. Using the TASE software, we found that 40 chromosomal regions were common to the two affected sisters, with an average length of 19 Mb (range: 5–87 Mb) and coverage of 78 SNPs/Mb (range: 1–129 SNPs/Mb; Table 2). The seven best QS (range: 461.7–369.7) were found to correspond (from highest to lowest QS) to regions of chromosomes 6, 5, 2, 1, 8, 6 and 2. In all, 8 of the 40 chromosomal regions contained 14 candidate genes previously reported in inherited retinal diseases. When comparing phenotypes caused by these candidate genes with the arRP of the two affected sisters, it was found that four genes, C2ORF71, ZNF513, PRCD and PDE6G were concordant. Of these, only C2ORF71 and ZNF513 were included in one of the seven chromosomal regions with the best QS, namely the chromosome two region being the seventh in QS rank. Among these two genes, C2ORF71 had previously been described in cone rod dystrophies,8, 9, 10 a phenotype corresponding closely to what was found in the two sisters of family RP1459. The exons of C2ORF71 were therefore sequenced in the five family members. We found two variants in exon 1; an insertion, c.2327_2328insC, and a deletion, c.2328_2344del17, both resulting in frameshifts, p.Leu777PhefsX34 and p.Leu777AsnfsX28, respectively (Figure 1d). These variants segregated with the RP phenotype (Figure 1c). Indeed, the two affected sisters carried both variants in trans whereas healthy sister and parents carried only one variant. These variants were therefore likely to be disease causing mutations.
Discussion
With the advent of clinical trials for inherited retinal dystrophies, it is required to identify the causative gene. Indeed, the molecular identification permits the diagnosis of the RP subtype, a better patient follow-up and prediction of disease course. This will also be necessary for gene therapy. However, molecular diagnosis in arRP, the most genetically heterogeneous form of inherited retinal disease, currently requires the screening of 32 genes, a process that was never fully completed by any research group because it is time and money consuming. To circumvent this difficulty, various genetic tests have been developed based on use of microchips testing for known mutations (Asper Biotech, Tartu, Estonia), use of re-sequencing microchips11 or preferential sequencing of mutation hot spots.12 Yet, these strategies miss unknown mutations and/or rare genes. Another possibility is to perform phenotype–genotype correlations to orient genetic testing towards one or a few genes. Although this could be very efficient in rare occasions in which a particular clinical feature is specific to a single gene, such as para-arteriolar preservation of the retinal pigment epithelium in CRB1 mutations,13 in most cases the RP phenotype simply belongs to a broad class of RP subtype, such as severe versus moderate, or rod–cone versus cone–rod dystrophy, all of which contain many genes to test.
Homozygosity mapping proved to be a very efficient method for identification of mutations and gene discovery in small inbred RP families,2, 14 as well as in about one-third of patients from outbred families who carry homozygous mutations.3, 4, 14 However, there remains about two-thirds of outbred families, being the most frequent in countries with highly mixed populations, in which affected patients carry compound heterozygous mutations. In these families, gene mapping could lead to the causative gene if there is a sufficient number of affected patients in the sibship, a rather rare occurrence today.15 Therefore, gene mapping of non-consanguineous families is usually used to ascertain a locus previously identified by homozygosity mapping, and to increase the probability of finding mutations in a novel gene.
We show in this study that combining gene mapping and phenotype–genotype correlation in small outbred families leads to the identification of the causative genes among several dozen of theoretically possible RP genes. Gene mapping is based on the assumption that the responsible gene must be present in chromosomal regions where affected patients from a single sibship have the same genotype. Although we found that in 260 000 SNP microchips many regions respond to this criterion, we could restrain the search to a few regions which had the highest QSs. The QS varies for the one part with the SNP coverage of the chromosomal region, and for the other part with the density of qualifying SNPs in the region, herein called the QR. Indeed, if the QR is low, this means that most SNPs of a given region have a genotype common to affected patients and to unaffected individuals of the kindred, making it unlikely to contain the causative gene. Conversely, if the QR is high, genotype of affected patients is different from that of unaffected individuals for many SNPs, therefore increasing the probability for the given region to contain the causative gene. With the increasing number of genes described in inherited retinal diseases, there are also an increasing number of precise phenotype descriptions.16 Thus, there were only a limited number of genes that fulfilled the criteria of a high QS and phenotype concordance. Therefore, combining both approaches appears useful for rapid mutation identification.
The TULP1 contains 542 amino acids, among which the ∼260 C-terminal amino acids form the tubby domain conserved in Tubby and in the three TULP proteins. TULP1 is specific to photoreceptor cells and is expressed in the inner segment, connecting cilium and synapses of photoreceptors. Today, 24 pathogenic variants have been described in TULP1, including 12 missense and 12 nonsense or frameshift mutations.17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27 In all, 10 out of the 12 missense mutations are present in the tubby domain. The two novel missense mutations, R311Q and R342Q, that we have found in this study also affect amino acids of the tubby domain. Using a protein data bank software (http://www.rcsb.org/pdb), we predict that Q311 would no longer be able to interact with F535 as does wild-type R311 do, therefore possibly destabilizing the protein. We also speculate that Q342, in contrast to wild-type R342, could increase the flexibility of the external loop of TULP1, therefore preventing protein–protein interactions. The early onset severe RP observed in the two patients carrying these mutations was in accordance with the phenotype described in TULP1 patients. Thus, R311Q and R342Q are likely pathogenic changes.
C2ORF71 encodes a 1288 amino acid protein with no known homology. The mRNA was shown to be specifically expressed in photoreceptors.8, 9 In these cells, C2ORF71 could possibly be associated with the photoreceptor connecting cilium and also have a role in the photoreceptor development, as it associates with basal bodies of the developing cilium.9 Using homozygosity mapping of consanguineous families, five pathogenic variants were recently reported in this gene,8, 9 four of them being nonsense or frameshift mutations. A medium scale systematic screening in 191 arRP and 95 Leber congenital amaurosis patients did not find any mutation, suggesting that C2ORF71 is not a frequent gene of inherited retinal dystrophies.10 The two novel variants found in this study are also frameshift mutations in compound heterozygous patients, unambiguously indicating that they are pathogenic. Among the five previously reported families, one Dutch family was described in more details, featuring criteria of cone rod dystrophy based on worse cone ERG responses when compared to rod ERG responses. This corresponds well to patient II.1 from this study, showing that phenotype–genotype correlation was relevant and, together with gene mapping, led to efficient mutation finding.
In conclusion, a combined approach of gene mapping and phenotypic assessment was efficient to find, among many possible chromosomal regions and causative genes, the mutations in two non-consanguineous families.
Accession codes
References
Hartong DT, Berson EL, Dryja TP : Retinitis pigmentosa. Lancet 2006; 368: 1795–1809.
den Hollander AI, Lopez I, Yzer S et al: Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest Ophthalmol Vis Sci 2007; 48: 5690–5698.
Hildebrandt F, Heeringa SF, Rüschendorf F et al: A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet 2009; 5: e1000353.
Harville HM, Held S, Diaz-Font A et al: Identification of 11 novel mutations in eight BBS genes by high-resolution homozygosity mapping. J Med Genet 2010; 47: 262–267.
Li L, Ho S, Chen C et al: Long contiguous stretches of homozygosity in the human genome. Hum Mutat 2006; 27: 1115–1121.
Marmor MF, Holder GE, Seeliger MW, Yamamoto S : Standard for clinical electroretinography (2004 update). Doc Ophthalmol 2004; 108: 107–114.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Collin RWJ, Safieh C, Littink KW et al: Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet 2010; 86: 783–788.
Nishimura DY, Baye LM, Perveen R et al: Discovery and functional analysis of a retinitis pigmentosa gene, C2ORF71. Am J Hum Genet 2010; 86: 686–695.
Sergouniotis PI, Li Z, Mackay DS et al: A survey of DNA variation of C2ORF71 in probands with progressive autosomal recessive retinal degeneration and controls. Invest Ophthalmol Vis Sci 2010. Available at http://www.ncbi.nlm.nih.gov/pubmed/20811058 (accédé 1 Février 1 2011).
Mandal MNA, Heckenlively JR, Burch T et al: Sequencing arrays for screening multiple genes associated with early-onset human retinal degenerations on a high-throughput platform. Invest Ophthalmol Vis Sci 2005; 46: 3355–3362.
Karra D, Jacobi FK, Broghammer M, Blin N, Pusch CM : Population haplotypes of exon ORF15 of the retinitis pigmentosa GTPase regulator gene in Germany : implications for screening for inherited retinal disorders. Mol Diagn Ther 2006; 10: 115–123.
den Hollander AI, ten Brink JB, de Kok YJ et al: Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat Genet 1999; 23: 217–221.
Littink KW, Koenekoop RK, van den Born LI et al: Homozygosity mapping in patients with cone-rod dystrophy: novel mutations and clinical characterizations. Invest Ophthalmol Vis Sci 2010; 51: 5943–5951.
Bergmann C, Senderek J, Anhuf D et al: Mutations in the gene encoding the Wnt-signaling component R-spondin 4 (RSPO4) cause autosomal recessive anonychia. Am J Hum Genet 2006; 79: 1105–1109.
Sparrow JR, Hicks D, Hamel CP : The retinal pigment epithelium in health and disease. Curr Mol Med 2010; 10: 802–823.
Gu S, Lennon A, Li Y et al: Tubby-like protein-1 mutations in autosomal recessive retinitis pigmentosa. Lancet 1998; 351: 1103–1104.
Hagstrom SA, North MA, Nishina PL, Berson EL, Dryja TP : Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with retinitis pigmentosa. Nat Genet 1998; 18: 174–176.
North MA, Naggert JK, Yan Y, Noben-Trauth K, Nishina PM : Molecular characterization of TUB, TULP1, and TULP2, members of the novel tubby gene family and their possible relation to ocular diseases. Proc Natl Acad Sci USA 1997; 94: 3128–3133.
Hanein S, Perrault I, Gerber S et al: Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat 2004; 23: 306–317.
Banerjee P, Kleyn PW, Knowles JA et al: TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nat Genet 1998; 18: 177–179.
den Hollander AI, van Lith-Verhoeven JJC, Arends ML, Strom TM, Cremers FPM, Hoyng CB : Novel compound heterozygous TULP1 mutations in a family with severe early-onset retinitis pigmentosa. Arch Ophthalmol 2007; 125: 932–935.
Kondo H, Qin M, Mizota A et al: A homozygosity-based search for mutations in patients with autosomal recessive retinitis pigmentosa, using microsatellite markers. Invest Ophthalmol Vis Sci 2004; 45: 4433–4439.
Mataftsi A, Schorderet DF, Chachoua L et al: Novel TULP1 mutation causing leber congenital amaurosis or early onset retinal degeneration. Invest Ophthalmol Vis Sci 2007; 48: 5160–5167.
Paloma E, Hjelmqvist L, Bayés M et al: Novel mutations in the TULP1 gene causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci 2000; 41: 656–659.
Lewis CA, Batlle IR, Batlle KG et al: Tubby-like protein 1 homozygous splice-site mutation causes early-onset severe retinal degeneration. Invest Ophthalmol Vis Sci 1999; 40: 2106–2114.
Abbasi AH, Garzozi HJ, Ben-Yosef T : A novel splice-site mutation of TULP1 underlies severe early-onset retinitis pigmentosa in a consanguineous Israeli Muslim Arab family. Mol Vis 2008; 14: 675–682.
Acknowledgements
We thank the patients and their family. This work was supported by private foundations (Fondation des Aveugles et Handicapés Visuels de France, Formicoeur, Information Recherche sur la Rétinite Pigmentaire, Retina France, SOS Rétinite and UNADEV), Centre National de Génotypage. INSERM and UNADEV support the fellowship for MH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Hebrard, M., Manes, G., Bocquet, B. et al. Combining gene mapping and phenotype assessment for fast mutation finding in non-consanguineous autosomal recessive retinitis pigmentosa families. Eur J Hum Genet 19, 1256–1263 (2011). https://doi.org/10.1038/ejhg.2011.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.133
Keywords
This article is cited by
-
Distinct mutations with different inheritance mode caused similar retinal dystrophies in one family: a demonstration of the importance of genetic annotations in complicated pedigrees
Journal of Translational Medicine (2018)
-
C2orf71a/pcare1 is important for photoreceptor outer segment morphogenesis and visual function in zebrafish
Scientific Reports (2018)
-
Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies
Scientific Reports (2016)
