
Abstract
Antisense-mediated exon skipping is a promising therapeutic approach for Duchenne muscular dystrophy (DMD) currently tested in clinical trials. The aim is to reframe dystrophin transcripts using antisense oligonucleotides (AONs). These hide an exon from the splicing machinery to induce exon skipping, restoration of the reading frame and generation of internally deleted, but partially functional proteins. It thus relies on the characteristic of the dystrophin protein, which has essential N- and C-terminal domains, whereas the central rod domain is largely redundant. This approach may also be applicable to limb-girdle muscular dystrophy type 2B (LGMD2B), Myoshi myopathy (MM) and distal myopathy with anterior tibial onset (DMAT), which are caused by mutations in the dysferlin-encoding DYSF gene. Dysferlin has a function in repairing muscle membrane damage. Dysferlin contains calcium-dependent C2 lipid binding (C2) domains and an essential transmembrane domain. However, mildly affected patients in whom one or a large number of DYSF exons were missing have been described, suggesting that internally deleted dysferlin proteins can be functional. Thus, exon skipping might also be applicable as a LGMD2B, MM and DMAT therapy. In this study we have analyzed the dysferlin protein domains and DYSF mutations and have described what exons are promising targets with regard to applicability and feasibility. We also show that DYSF exon skipping seems to be as straightforward as DMD exon skipping, as AONs to induce efficient skipping of four DYSF exons were readily identified.
Similar content being viewed by others

Introduction
Limb-girdle muscular dystrophy type 2B (LGMD2B), Myoshi myopathy (MM) and distal myopathy with anterior tibial onset (DMAT) are autosomal recessive allelic muscle diseases caused by mutations in the dysferlin-encoding DYSF gene, leading to severely reduced or complete absence of the dysferlin protein.1, 2, 3 Most patients have small mutations: stop- or frameshift mutations lead to prematurely truncated proteins, whereas missense mutations generally affect protein stability.4 Over 100 different mutations have been reported in the Leiden Open Variation Database for almost 200 patients (www.dmd.nl). As there is currently no treatment for the ‘dysferlinopathies’, lack of dysferlin leads to progressive loss of tissue and function in the muscles of the limbs and girdle.5
The dysferlin protein is expressed in many tissues, but most abundantly in the heart and skeletal muscle.5 In the latter, the protein is located at the plasma membrane and in cytoplasmic vesicles.6 It is thought that dysferlin has a function in vesicle trafficking and membrane patch fusion repair in muscle cells.5 Loss of dysferlin compromises skeletal muscle membrane repair and leads to progressive loss of muscle fibers.6 The protein has different domains (Figure 1). The ENSEMBL database predicts six or seven calcium-dependent C2 lipid-binding (C2) domains, a transmembrane domain and multiple ‘fer’ and ‘dysf’ domains. The C2 domains probably mediate calcium-dependent vesicle fusion with the plasma membrane, whereas the transmembrane domains anchor the protein to the plasma membrane.5 The fer and dysf domains have as yet an unknown function.4

Dysferlin domains relative to DYSF exons. Dysferlin contains six or seven calcium-dependent C2 lipid binding domains (C2), a transmembrane domain (T), a ferl domain (L), FerA and FerB domains (A and B, respectively) and Dysf_N and Dysf_C domains (N and C, respectively). The C2 and transmembrane domains have a function in membrane repair. The function of other domains is yet unknown.
It is likely that parts of the dysferlin protein are redundant. The first indication for this is a finding by Sinnreich et al7 that the mother of two LGMD2B patients turned out to be a compound heterozygote rather than a carrier. One of the alleles contained a mutated branchpoint in intron 31, leading to skipping of exon 32. As exon 32 skipping does not disrupt the open reading frame, this resulted in a slightly shorter but apparently partly functional dysferlin protein at levels that were 10% of wild-type levels. The patient had only very mild proximal muscle weakness, elevated serum creatine kinase levels and was still ambulant at age 70. In contrast, her severely affected daughters were homozygous for a null mutation and had no dysferlin protein. In addition, a mildly affected patient has been presented with a dysferlin containing only the final two C2 and the transmembrane domains.8 This patient was ambulant without a cane at age 41. Further proof for the functionality of this protein came from its proper location at the sarcolemma and the delivery of a gene encoding this ‘minidysferlin’ into a dysferlin-negative mouse model through an adeno-associated viral vector. This resulted in detectable levels of the mini-dysferlin protein and an improved phenotype.
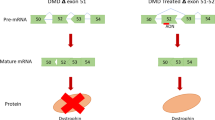
Thus, it is possible that bypassing dysferlin mutations could lead to more stable, (partly) functional dysferlin proteins and would therefore have therapeutic potential. A way to achieve this is the modulation of dysferlin pre-mRNA splicing using antisense oligonucleotides (AONs) or antisense sequences, which hide target exons from the splicing machinery, such that they are not included into the final mRNA (‘exon skipping’) (Figure 2). Initial proof of concept was recently shown in control and patient-derived cells, were exon 32 skipping was induced by either AONs or through an a modified U7 snRNP in which the original antisense sequence was replaced by the target antisense sequence.9

Antisense-mediated exon skipping. Left panel: in this example, a mutation within exon 32 results in a premature stop codon (indicated by the transition of black to white in the pre-mRNA (top) and mRNA (middle), which leads to a prematurely truncated protein (bottom). Right panel: when antisense oligonucleotides (AON) targeting exon 32 are used, they will hybridize to this exon, thus hiding it from the splicing machinery, resulting in the skipping of this exon. As exon 32 in in-frame (its length is divisible by three), skipping will not disrupt the reading frame (the mRNA becomes black in the middle panel) and a full-length protein, which misses a little bit in the middle, can be generated (bottom).
In fact, the exon skipping approach is currently the most promising therapeutic approach for Duchenne muscular dystrophy (DMD).10 This disease is caused by a complete loss of functional dystrophin due to mutations that disrupt the open reading frame of the DMD gene.11 Similar to dysferlin, dystrophin contains essential and less essential domains, underlined by the finding that patients with in-frame mutations in the redundant central domain suffer from a milder disease called Becker muscular dystrophy.12, 13, 14 AON-medicated exon skipping for DMD patients aims to restore the open reading frame, to allow the production of a partly functional protein and conversion of the severe DMD into a milder BMD phenotype. This approach was pioneered in our lab and by colleagues in Japan, Australia and the United Kingdom10, 15, 16, 17, 18 and is currently in phase I/II clinical trials.19, 20
In this study we will analyze what DYSF exons might be suitable targets for antisense-mediated exon skipping and what are less or nonsuitable targets, based on the protein encoding domains and reported mutations. We also show that using our guidelines established after retrospective analysis of our set of DMD AONs, AONs to induce DYSF exon skipping can be readily identified.
Methods
Database
Mutations as reported on 8 May 2009 in the DYSF LOVD database (www.dmd.nl) were analyzed.
AONs
AON design was based on our guidelines for DMD exons and focused on targeting partially open secondary RNA structures (predicated by m-fold21), the presence of predicted RESCUE-ESE and SC35 and the absence of predicted Tra2β sites (using the human splicing finder22) and favorable binding energy.21, 23 All AONs (Table 1) target exon-internal sequences and consist of 2′-O-methyl RNA with a full-length phosphorothioate backbone and were manufactured by Eurogentec (Seraing, Belgium).
Cell culture and transfection
Human control myoblasts were cultured and differentiated as described previously.24 AONs were transfected at a 500 nM concentration, using 2.5 μl polyethyleneimine (MBI-Fermentas, Leon-Rot, Germany) per μg AON according to the manufacturer's instructions. An unrelated 5-fluorescein-labeled AON targeting exon 45 of the dystrophin gene was used to confirm the efficiency of transfection (>90%).
RNA analysis
RNA was isolated at >28 h after transfection using RNA-Bee (Campro Scientific, Veenendaal, the Netherlands) according to the manufacturer's instruction. An RT-PCR was performed with random hexamer primers, as described.25 Primers flanking the targeted exons (sequence on request) were used to amplify the cDNA as described previously for dystrophin,24 but using a single PCR for 35 cycles. Skip products were analyzed by sequencing analysis as described.24
Results and discussion
Exon skipping for dysferlin
Obviously, not every DYSF exon can be skipped without consequence for dysferlin function. First, if the skipped exon is out-of-frame (ie, the length is not divisible by three), this will result in a disruption of the open reading frame and a prematurely truncated protein. Thus, either in-frame exons, or a combination of out-of-frame exons that together maintain the reading frame, are valid targets (Figure 3 and Table 2).

Dysferlin exons. In-frame exons are depicted in white, out-of-frame exons in black. Exons or combinations of exons can be skipped without disrupting the reading frame, when the resulting ends fit (eg, exons 39 and 40 can be skipped, as the end of exon 38 fits to the beginning of exon 41).
Second, as mentioned, dysferlin contains several domains, and although only limited information is available about their function and essentiality, several things can be learned about these domains from mutations found in patients and animal models. The very mildly affected individual skipping exon 32 suggests that, although exon 32 encodes the fourth C2 domain, a dysferlin without this exon is highly functional.7 Thus, apparently the fourth C2 domain is (at least partially) redundant. In contrast, the final C2 domain is likely essential for functionality, as a mouse model SJL/J (SJL-Dysf) with a splice site mutation resulting in the in-frame skipping of exon 45, leading to the omission of the last part of the final C2 domain, has a dystrophic phenotype.26, 27 The mild patient producing a dysferlin consisting of only the last C2 and the transmembrane domains could suggest that the other four C2 domains are redundant. However, whereas this corresponding mini-dysferlin protein is apparently relatively stable, this does not necessarily hold for all dysferlins with mutations before exon 44, and seems to be an exception rather than a rule.4
Third, only internal exons can be skipped, and thus exons 1 and 55 are invalid targets. Additional exons that provide invalid targets are in-frame exons 19, 25 and 49 for which splice site mutations resulting in exon skipping (confirmed by RT-PCR) have been found in LGMD2B and MM patients (4 and the LOVD DYSF database). Mutations that may affect splicing (ie, located at or close to the splice sites) have been identified in MM and LGMD2B patients for exons 24, 30, 32, 34, 37 and 41. However, the mutations were found at the DNA level and have not been confirmed at the RNA level.
On the basis of this information, exons can be subdivided into suitable and less suitable and impossible candidates (Table 2). Although there are no real mutational hotspots in the DYSF gene, some exons contain more mutations than others and the skipping of these exons would thus be applicable to larger groups of patients (see Table 2). Notably, no mutations have been thus far reported for exons 17 and 35.
Prioritizing DYSF exons
Obviously, when assessing the feasibility of DYSF exon skipping, one should start with the most straightforward approach (single exon skipping), targeting those exons that are most likely or have been confirmed to be redundant. This includes exons 17, 32, 35, 36 and 42. As no mutations have been reported yet for exons 17 and 35, initial focus should not be on those exons. Exons 34 and 41 are in-frame and theoretically eligible targets as they encode no domains or the redundant C2 domain. However, putatively causal mutations that induce skipping of these exons have been identified at the DNA level. Thus, exons 32 and 36 seem to be prime candidates, with the other mentioned exons close seconds.
As skipping two or three exons simultaneously (double and triple exon skipping, respectively) is feasible for DMD,25, 28 it is probably also feasible for DYSF. The only combination of adjacent exons that together is in-frame (ie, the total number of nucleotides is divisible by three) while not encoding any domains is 20+21, whereas there are also some in-frame three exon combinations possible (18+19+20 and 31+32+33).
Finally, in-frame exons 24 and 30 encode the FerB and Dysf-C2 domains, the additional domains for which the essentiality for dysferlin function is still unknown. Thus, exon skipping for these exons might be beneficial. All other exons or exon combinations encode C2 domains or the transmembrane domain and are very likely to interfere with protein function, and should have a lower priority (Table 2).
Skipping the highest priority exons (rank 1–3) could be beneficial for over 20% of all dysferlinopathy patients (Table 2). This is probably a slight overestimation, as a number of patients may be counted twice as they have mutations in two eligible exons (compound heterozygotes). The recessive nature of dysferlinopathies poses certain obstacles that are not present in the X-recessive DMD. For instance, a patient can have mutations in exons 32 and 30. When targeting both exons, it is likely that in most transcripts both exons will be skipped, as double exon skipping is usually quite efficient.25 This is probably not beneficial. However, levels of ∼10% are apparently sufficient to prevent disease in the aforementioned patient who lacked exon 32,7 suggesting that it is not essential to correct both mutations and/or restore dysferlin levels to 100%. Therefore, it may be better to target just one mutated exon (eg, exon 32), which will result in skipping of the unintended exon (wild-type exon 32 in the exon 30 mutated transcript), but also of the intended exon (mutated exon 32). The intended exon skipping will result in functional dysferlin, whereas the unintended exon skipping will not further worsen the disease as a non-functional dysferlin (due to the exon 30 mutation) is now converted in another non-functional dysferlin (exon 30 mutation and exon 32 skip).
Feasibility of DYSF exon skipping
Antisense-mediated skipping of DMD exons is very straightforward; AONs to skip each exon have been identified.23, 29, 30 To assess whether exon skipping can be as easily achieved for DYSF exons, we designed two AONs for each exon targeting DYSF exons 19, 24, 30 and 34, and a single AON targeting the small exon 32, using our previously identified AON design guidelines.23, 29 AONs were transfected in differentiated human control myoblasts. RT-PCR analysis revealed that six AONs were effective and induced skipping of exons 19, 24, 30 and 34 (Figure 4). Exon skipping levels varied from 17% (exon 34) to 96% (exon 30). Notably, some spontaneous skipping (alternative splicing) of exon 30 was observed in non-treated cells at low levels (10%). The single exon 32 AON was not effective (Figure 4), but exon 32 skipping can be induced by other AONs.9

RT-PCR analysis of AON-treated control cell cultures. AONs 19–2, 24–1, 24–2, 30–1, 30–2 and 34–1 are effective, whereas 19–1, 32, 34–2 and C (a control AON targeting the DMD (dystrophin) gene) are not. Correct exon skipping was confirmed using sequence analysis (data not shown). No exon 19, 24, 32 or 34 skipping could be observed in nontreated (NT) cells, whereas for exon 30 low levels of physiological skipping were observed. AON treatment significantly increased these levels from <10 to >90%. Skipping percentages using Lab-on-a-chip (Agilent, Amstelveen, the Netherlands) are indicated below each skip. Note that the intensity of the skip products is lower, due to the smaller fragment length (our efficiency assessment corrects for this). –RT and H2O are negative controls. M is size marker.
For some DMD exons, skipping of additional adjacent exons was observed, either exclusively or in addition to skipping of the targeted exon only.29 This is unwanted for dysferlinopathies as the skipping of a number of in-frame exons leads to non-functional dysferlins (Table 2). However, skipping of additional exons was not observed for the four dysferlin exons targeted in this study (data not shown). Furthermore, the skipping of multiple exons happens only occasionally for DMD exons and even when it occurs the amount of transcripts with a single skip often vastly exceeds the amount of transcripts in which more exons are skipped consecutively.
Concluding remarks
In conclusion, exon skipping poses a putative strategy for dysferlinopathies, although some exons are arguably more eligible than others. It should be pointed out that the ideal target mutations are homozygous mutations in an eligible target exon, as in this case skipping the targeted exons on both alleles could be therapeutic. However, as 10% of dysferlin levels seem to be sufficient to improve the phenotype,7 this approach is also promising for heterozygous mutations. Skipping of only the mutated exon here could be potentially therapeutic, whereas skipping of the non-target allele is not. Nevertheless, this could still lead to dysferlin levels of up to 50%. Fortunately, skipping DYSF exons seems to be straightforward. It is known that some mutations can cause misfolding of dysferlin and retention in the endoplasmatic reticulum.31 This is also a possibility for the internally deleted dysferlins. Therefore, studies in patient-derived cells are needed, as only these will be able to confirm whether other internally deleted dysferlins are functional and located properly.
References
Liu J, Aoki M, Illa I et al: Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 1998; 20: 31–36.
Bashir R, Britton S, Strachan T et al: A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet 1998; 20: 37–42.
Illa I, Serrano-Munuera C, Gallardo E et al: Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol 2001; 49: 130–134.
Therrien C, Dodig D, Karpati G, Sinnreich M : Mutation impact on dysferlin inferred from database analysis and computer-based structural predictions. J Neurol Sci 2006; 250: 71–78.
Bansal D, Campbell KP : Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol 2004; 14: 206–213.
Bansal D, Miyake K, Vogel SS et al: Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003; 423: 168–172.
Sinnreich M, Therrien C, Karpati G : Lariat branch point mutation in the dysferlin gene with mild limb-girdle muscular dystrophy. Neurology 2006; 66: 1114–1116.
Krahn M, Wein N, Lostal W et al: Partial functionality of a mini-dysferlin molecule identified in a patient affected with moderately severe primary dysferlinopathy. Neuromuscul Disord 2008; 18: 781.
Wein N, Avril A, Bartoli M et al: Efficient bypass of mutations in dysferlin deficient patient cells by antisense-induced exon skipping. Hum Mutat 2009, e-pub ahead of print 1 December 2009.
van Ommen GJ, van Deutekom J, Aartsma-Rus A : The therapeutic potential of antisense-mediated exon skipping. Curr Opin Mol Ther 2008; 10: 140–149.
Hoffman EP, Brown Jr RH, Kunkel LM : Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987; 51: 919–928.
Hoffman EP, Fischbeck KH, Brown RH et al: Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy. N Engl J Med 1988; 318: 1363–1368.
Koenig M, Monaco AP, Kunkel LM : The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988; 53: 219–226.
Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM : An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988; 2: 90–95.
Dunckley MG, Manoharan M, Villiet P, Eperon IC, Dickson G : Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum Mol Genet 1998; 7: 1083–1090.
Takeshima Y, Wada H, Yagi M et al: Oligonucleotides against a splicing enhancer sequence led to dystrophin production in muscle cells from a Duchenne muscular dystrophy patient. Brain Dev 2001; 23: 788–790.
Wilton SD, Lloyd F, Carville K et al: Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul Disord 1999; 9: 330–338.
van Deutekom JC, Bremmer-Bout M, Janson AA et al: Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum Mol Genet 2001; 10: 1547–1554.
Muntoni F, Bushby KD, van Ommen G : 149th ENMC international workshop and 1st TREAT-NMD workshop on: ‘planning phase i/ii clinical trials using systemically delivered antisense oligonucleotides in Duchenne muscular dystrophy’. Neuromuscul Disord 2008; 18: 268–275.
van Deutekom JC, Janson AA, Ginjaar IB et al: Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med 2007; 357: 2677–2686.
Zuker M : Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31: 3406–3415.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C : Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009; 37: e67.
Aartsma-Rus A, van Vliet L, Hirschi M et al: Guidelines for antisense oligonucleotide design and insight into splice-modulating mechanisms. Mol Ther 2008.
Aartsma-Rus A, Janson AA, Kaman WE et al: Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum Mol Genet 2003; 12: 907–914.
Aartsma-Rus A, Janson AA, Kaman WE et al: Antisense-induced multiexon skipping for duchenne muscular dystrophy makes more sense. Am J Hum Genet 2004; 74: 83–92.
Vafiadaki E, Reis A, Keers S et al: Cloning of the mouse dysferlin gene and genomic characterization of the SJL-Dysf mutation. NeuroReport 2001; 12: 625–629.
Bittner RE, Anderson LV, Burkhardt E et al: Dysferlin deletion in SJL mice (SJL-Dysf) defines a natural model for limb girdle muscular dystrophy 2B. Nat Genet 1999; 23: 141–142.
Aartsma-Rus A, Janson AA, van Ommen GJ, van Deutekom JC : Antisense-induced exon skipping for duplications in Duchenne muscular dystrophy. BMC Med Genet 2007; 8: 43.
Aartsma-Rus A, Winter CL, Janson AAM et al: Functional analysis of 114 exon-internal AONs for targeted DMD exon skipping: indication for steric hindrance of SR protein binding sites. Oligonucleotides 2005; 15: 284–297.
Wilton SD, Fall AM, Harding PL, McClorey G, Coleman C, Fletcher S : Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol Ther 2007; 15: 1288–1296.
Fujita E, Kouroku Y, Isoai A et al: Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum Mol Genet 2007; 16: 618–629.
Acknowledgements
AAR is funded by grants from ZonMw (The Netherlands) and the Duchenne Parent Project (The Netherlands) and the FP6 TREAT-NMD network of excellence.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
AA-R and G-JBvO are employees of Leiden University Medical Center and coinventors on patent applications for antisense sequences and exon-skipping technology.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Aartsma-Rus, A., Singh, K., Fokkema, I. et al. Therapeutic exon skipping for dysferlinopathies?. Eur J Hum Genet 18, 889–894 (2010). https://doi.org/10.1038/ejhg.2010.4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.4
Keywords
This article is cited by
-
Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy
Gene Therapy (2020)
-
Gene therapy matures in the clinic
Nature Biotechnology (2012)
-
Therapeutic exon ‘switching’ for dysferlinopathies?
European Journal of Human Genetics (2010)
