
Abstract
Imputation is an extremely valuable tool in conducting and synthesising genome-wide association studies (GWASs). Directly typed SNP quality control (QC) is thought to affect imputation quality. It is, therefore, common practise to use quality-controlled (QCed) data as an input for imputing genotypes. This study aims to determine the effect of commonly applied QC steps on imputation outcomes. We performed several iterations of imputing SNPs across chromosome 22 in a dataset consisting of 3177 samples with Illumina 610k (Illumina, San Diego, CA, USA) GWAS data, applying different QC steps each time. The imputed genotypes were compared with the directly typed genotypes. In addition, we investigated the correlation between alternatively QCed data. We also applied a series of post-imputation QC steps balancing elimination of poorly imputed SNPs and information loss. We found that the difference between the unQCed data and the fully QCed data on imputation outcome was minimal. Our study shows that imputation of common variants is generally very accurate and robust to GWAS QC, which is not a major factor affecting imputation outcome. A minority of common-frequency SNPs with particular properties cannot be accurately imputed regardless of QC stringency. These findings may not generalise to the imputation of low frequency and rare variants.
Similar content being viewed by others

Introduction
Genome-wide association scans (GWASs) have proven to be a successful strategy for detecting common variants exerting modest effects on complex disease risk. Currently available commercial platforms focus on common variants and capture the majority of HapMap1 SNPs with minor allele frequency (MAF) >0.05 in European populations.2 Several large-scale consortia have been formed in order to carry out GWAS meta-analyses for various phenotypes, with successful outcome (eg, Zeggini et al,3 Prokopenko et al,4 Franke et al,5 Barret et al6 and Soranzo et al7). To enable the combination of data across studies carried out on different platforms, and to enable in silico fine mapping of association signals, imputation approaches were proposed a few years ago8 as a means of statistically inferring genotypes at untyped loci using a reference set, for example, the HapMap (∼2 500 000 SNPs).
An important aspect of any GWAS analysis is the implementation of a series of rigorous quality control (QC) steps before testing for association. These QC procedures help guard against genotyping error, population stratification, sample duplication and other confounders that can affect the analysis results. QC steps are typically applied at the sample- and SNP-specific level. Sample-level QC includes filtering out samples with low call rates, evidence for different ethnic origin, high heterozygosity, relatedness/duplication, gender discrepancies and genotyping batch effects. SNP-level QC includes filtering out SNPs with low call rates and deviation from Hardy–Weinberg equilibrium (HWE) at pre-determined thresholds. It is generally believed that datasets should be stringently quality controlled (QCed) at the marker level before applying imputation approaches. For this reason, lower MAF SNPs tend to also be excluded, as their accuracy can be hampered by poor clustering properties and incorrect automated genotype calling (at least with currently widely used algorithms). Even though such weight is placed on pre-imputation SNP QC, the effects of applying different criteria and thresholds to the starting dataset have not been investigated thus far. In this report, we evaluate the effect of GWAS QC on imputation outcome, and find that imputation works very well for common variants irrespective of QC, and that a minority of some common-frequency SNPs with particular properties cannot be accurately imputed regardless of QC stringency.
Materials and methods
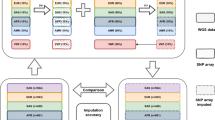
We used an empirical GWAS dataset to assess the effect of QC on imputation outcome. We focused on chromosome 22, (n=9038 directly typed SNPs) from 3177 osteoarthritis (OA) cases from the United Kingdom, typed on the Illumina 610k quad chip (Illumina) as part of the arcOGEN consortium GWAS (manuscript submitted). Chromosome 22 is representative of the genome in terms of the proportion of directly typed to imputed SNPs. All samples included in our analysis had passed standard sample level QC (based on call rate, heterozygosity, relatedness, ethnicity and gender discrepancies). We imputed genotypes at variants on the basis of HapMap phase II release 22 CEU data (n=33 815 SNPs on chr22) using IMPUTE v1 (https://mathgen.stats.ox.ac.uk/impute/impute.html).8 We performed each imputation in duplicate, with and without the IMPUTE v1 predict genotyped SNPs flag, which resulted in one set of imputed data containing the original genotypes and in the other imputed genotypes. To assess the effect of varying levels of QC, we carried out several rounds of imputation, using differently QCed OA SNP data as the starting point.
Initially, we imputed on the basis of no SNP-level QC, including all directly typed SNPs, regardless of MAF, call rate and HWE. We also imputed on the basis of only those SNPs that passed stringent QC thresholds (call rate >95% for SNPs with a MAF ≥5% and call rate >99% for SNPs with a MAF <5%, HWE exact P>0.0001, MAF >0.01 and removing all SNPs with GC or TA alleles; Table 1). Although imputation biases can occur due to poor clustering of SNPs with miscalled genotypes in the starting dataset, cluster plot checking is not feasible at the genome-wide scale and therefore, it is not implemented in standard GWAS QC.
We evaluated the accuracy of imputed genotypes by comparing allele frequencies at the same SNP between imputed and true, directly typed data. For each QC-imputation iteration, we performed an allele frequency comparison between the actual directly typed and imputed SNPs. Under perfect imputation, we would expect to see alignment with the null hypothesis of no association. We used SNPTEST (http://www.stats.ox.ac.uk/~marchini/software/gwas/snptest.html)9 to investigate differences between directly typed and imputed genotypes at the same variants within the same samples, taking into account the distribution of genotype probabilities for each individual. For the purposes of our comparison, we used those SNPs that were directly genotyped in OA cases and also present in the HapMap reference samples. Table 1 summarises the number of these SNPs for each QC threshold.
When comparing directly typed with imputed allele frequencies at the same variant in the same individuals, we arbitrarily considered P<10−6 as significantly different. We calculated the correlation between imputed and directly typed MAF, using the expected counts to allow for genotype-associated probabilities. We also applied a series of post-imputation QC steps in order to eliminate unreliably imputed SNPs, aiming to filter out as many of these SNPs as possible while retaining a good proportion of nonsignificant SNPs. We compared two alternative methods for post-imputation QC filtering, first, the IMPUTE-info score, which is associated with the imputed allele frequency estimate which ranges from 1, indicating high confidence, to 0 suggesting decreased confidence, and second, the freq-add-proper-info score provided by SNPTEST, a relative statistical score ranging from 0 to 1, representing no information to complete information, respectively. The SNPTEST freq-add-proper-info score has been shown to be highly correlated with the IMPUTE-info score under the additive model.10 In both scenarios, we also filtered out SNPs with MAF <5%. Figure 1 illustrates the effects of altering post-imputation QC filters on the QCed data. On the basis of these results, we chose to use the IMPUTE-info score with a filtering threshold <0.8 and MAF <5%, which effectively eliminated ∼79% of the significant SNPs while retaining ∼85% of the nonsignificant ones (SNPTEST freq-add-proper-info <0.9 and MAF 5% would be roughly equivalent to this eliminating ∼73% of the significant SNPs while retaining ∼89% of the nonsignificant ones). We applied this post-imputation filter to each of our datasets and compared the results. We looked at the unQCed and QCed datasets first, as synopsised in Table 1. For each scenario, we examined frequency differences between the directly typed and the imputed genotypes as described above. In addition, we compared the imputed genotypes at imputed SNPs only for the unQCed and the fully QCed (QCed data with all poorly clustered markers removed) strategies.

(a) Imputation results for the QCed data indicating the total number of SNPs filtered for different QC thresholds using the IMPUTE-info and freq-add-proper-info scores. The SNPs remaining after the filter (red bar) have been subdivided into SNPs that are significant (green bar) and not significant (yellow bar). (b) The same data as percentage of significant and nonsignificant SNPs removed for each threshold. Both methods of filtering appear to be equivalent, but the freq-add-proper-info is shifted to the right for the same numerical threshold; we chose the IMPUTE-info <0.8 for further analysis (similar to a freq-add-proper-info <0.9).
Results
Table 1 summarises the number of SNPs with significantly (P<10−6) different allele frequencies between the directly typed and imputed data in the same set of individuals for each of the different QC sets. Correlation plots and R2 values for the comparisons of the QCed and unQCed datasets are presented in Figure 2. The difference between the unQCed (R2=0.993) and QCed data (R2=0.994) was minimal. After post-imputation filtering there were 77 SNPs with significantly different (imputed v. directly typed) allele frequencies in the unQCed data compared with 67 significant SNPs in the QCed data. In an attempt to improve imputation for the small subset of poorly imputed SNPs in the QCed data, we excluded all SNPs with MAF<5% and, subsequently, also SNPs with MAF<10%. We found that eliminating these lower MAF SNPs before imputation had little effect overall. The R2 for the post-imputation QC filtered comparison with the QCed data was virtually identical both when excluding all SNPs with MAF<5% (R2=0.994) and when excluding all SNPs with MAF<10% (R2=0.991).

Correlation plots and the associated R2 for (a) The unQCed and the QCed with and without post-imputation QC filtering (IMPUTE-info <0.8 and MAF <5%). (b) The imputed-only markers in the unQCed and fully QCed data (QCed data with all poorly clustered markers removed) without post-imputation QC filtering.
Given this apparent minimal influence of input data QC on imputation outcome, we investigated further the small set of SNPs showing significant allele frequency differences for the presence of a common characteristic that could conceivably be used as a post-imputation filter. To rule out poor genotyping as the cause of these significant differences, we examined all cluster plots for the unfiltered significant SNPs (P<1 × 10−6, n=325). In all, 14 poorly clustered SNPs were removed and the data were re-imputed. After post-imputation QC, three additional SNPs were not significant and six were less significant. We then inspected the cluster plots for 10 SNPs on either side of the 61 SNPs remaining significantly different to rule out poor imputation due to flanking SNP poor clustering properties. We examined the cluster plots for 1008 SNPs and found that 36 of these were poor; these resided in the proximity of 35 of the significant SNPs. We subsequently removed these SNPs and re-imputed. We found that following post-imputation QC filtering, only 3 of the 61 SNPs were no longer significant, and the R2 remained the same as for the QCed data (R2=0.994) for the post-imputation QC filtered data. When we repeated comparisons using IMPUTE v2 with the HapMap3 (CEU, release no. 2 February 2009) and data from the 1000 genomes project (Pilot 1 genotypes released March 2010; phased haplotypes released June 2010) as the reference panels, we observed qualitatively similar results.
Differences in region-specific recombination rates may account for the few remaining significant SNPs, as variants in areas of especially high recombination rate may be more challenging to impute accurately regardless of QC. To investigate this, we first examined the QCed unfiltered data and found that when the data were dichotomised into those markers with lower (<1 cm/Mb) and higher (≥1 cM/Mb) recombination rates, there were more significant SNPs present in the higher recombination rate group compared with the lower recombination group (P=1.85 × 10−27, average recombination rates of 12.8 and 3.04, respectively). When we examined the QCed data post-imputation QC, this difference disappeared (P=0.526). This clearly indicates that application of the post-imputation QC filter successfully identifies the majority of significant SNPs with high recombination rates. Therefore, to include recombination rate as an extra filter would not be prudent, for example, using the QCed post-imputation QC filtered data and applying a further filter using a recombination rate threshold of >1 cM/Mb would eliminate 2075 SNPs, only 24 of which are significantly different.
Discussion
The imputation accuracy of common variants does not appear to be substantially affected by GWAS QC steps. Our data demonstrate that there is little difference in imputation accuracy observed in unQCed GWAS data when compared with QCed GWAS data. Furthermore, the implementation of additional QC steps (eg, filtering out variants with MAF<0.05 and <0.10) does not considerably improve overall imputation accuracy. Missing variants and directly typed variants that fail pre-imputation QC checks are imputed and these data are used for downstream analyses. Post-imputation QC successfully eliminates a good proportion of inaccurately imputed SNPs. Specifically, by applying a very stringent post-imputation QC threshold, a smaller set of variants with more accurately predicted genotypes remain. The IMPUTE-info threshold of <0.8 and MAF ≤5% criterion successfully filtered out the majority of poorly imputed SNPs. However, the application of these strict filters in GWAS data could result in many SNPs being excluded from the data, and thus potential true association signals could be missed. Some of the inaccurately imputed variants were due to poor clustering properties. It is plausible that the handful of variants that still remained inaccurately imputed could be because of the differences in ethnicity between our data and the HapMap CEU reference panel from which the genotypes were predicted. We have used IMPUTE, but do not expect our results and conclusions to qualitatively differ with different imputation methods, for example, BEAGLE and MACH exhibit similar imputation accuracy to IMPUTE.11 Differences in population structure between the reference panel and target dataset can be a source of imputation inaccuracy. Imputation accuracy for common SNPs may be further increased by using larger reference panels with data on denser sets of variants. Our results show that GWAS QC is not of paramount importance for the imputation of common variants. This may be different for the imputation of low frequency and rare variants based on emerging reference panels such as the 1000 genomes (http://www.1000genomes.org) and UK10k (http://www.uk10k.org) projects. In summary, our study demonstrates that imputation of common variants is generally very accurate and robust to GWAS QC, which is not a major factor affecting imputation outcome.
References
The International HapMap Consortium: The International HapMap Project. Nature 2003; 426: 789–796.
Barrett JC, Cardon LR : Evaluating coverage of genome-wide association studies. Nat Genet 2006; 38: 659–662.
Zeggini E, Scott LJ, Saxena R et al: Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 2008; 40: 638–645.
Prokopenko I, Langenberg C, Florez JC et al: Variants in MTNR1B influence fasting glucose levels. Nat Genet 2009; 41: 77–81.
Franke A, Balschun T, Karlsen TH et al: Replication of signals from recent studies of Crohn's disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet 2008; 40: 713–715.
Barret JC, Clayton DG, Concannon P et al: Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009; 41: 703–707.
Soranzo N, Spector TD, Mangino M et al: A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet 2009; 41: 1182–1190.
Marchini J, Howie B, Myers S et al: A new multipoint method for genome-wide association studies via imputation of genotypes. Nat Genet 2007; 39: 906–913.
The Wellcome Trust Case Control Consortium: Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 2007; 447: 661–678.
Marchini J, Howie B : Genotype imputation for genome-wide association studies. Nat Rev Genet 2010; 11: 499–511.
Nothnagel M, Ellinghaus D, Schreiber S et al: A comprehensive evaluation of SNP genotype imputation. Hum Genet 2009; 125: 163–171.
Acknowledgements
EZ is supported by the Wellcome Trust (WT088885/Z/09/Z), LS is supported by the European Community Framework seven large collaborative project grant TREAT-OA, KC is supported by a Botnar Fellowship and by the Wellcome Trust (WT079557MA), NWR is supported by the Wellcome Trust (WT079557MA), JMW is supported by the Higher Education Funding Council for England. JL receives support from the United Kingdom NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Hospitals NHS Foundation Trust. The arcOGEN consortium is funded by a special purpose grant from Arthritis Research UK (Grant 18030).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Southam, L., Panoutsopoulou, K., Rayner, N. et al. The effect of genome-wide association scan quality control on imputation outcome for common variants. Eur J Hum Genet 19, 610–614 (2011). https://doi.org/10.1038/ejhg.2010.242
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.242
Keywords
This article is cited by
-
Impact of pre- and post-variant filtration strategies on imputation
Scientific Reports (2021)
-
Validation of genotype imputation in Southeast Asian populations and the effect of single nucleotide polymorphism annotation on imputation outcome
BMC Medical Genetics (2018)
-
Genome-wide association study identifies a new susceptibility locus for cleft lip with or without a cleft palate
Nature Communications (2015)
-
Conjunctival fibrosis and the innate barriers to Chlamydia trachomatis intracellular infection: a genome wide association study
Scientific Reports (2015)
-
Predicting HLA genotypes using unphased and flanking single-nucleotide polymorphisms in Han Chinese population
BMC Genomics (2014)
