
Abstract
Partial monosomy 21 was recently segregated into three regions associated with variable clinical severity. We describe 10 new patients, all examined by single nucleotide polymorphism (SNP) genotyping and G-banded karyotyping. Cohort A consisted of three patients seen in our medical genetics clinics with partial chromosome 21 monosomies. In two of these patients having terminal deletions (21q22.2-ter and 21q22.3-ter), the breakpoints differed by at least 812 Kb of sequence, containing seven RefSeq genes. A third patient had an interstitial hemizygous loss of 16.4 Mb (21q21.1–q22.11). All three patients had relatively mild phenotypes. Cohort B consisted of seven patients with partial chromosome 21 monosomies who had a greater number of dysmorphic features and some major malformations; SNP genotypes were obtained from the Coriell Genetic Cell Repository. We also collected data on partial monsomy 21 cases from the DECIPHER database. This report of 10 new cases of 21q deletion and review of a total of 36 confirms that deletion of the terminal region is associated with a mild phenotype, but suggests that deletion of regions 1 and 2 is compatible with life and have a variable phenotype perhaps relating more to other genetic and environmental variables than to genes in the interval.
Similar content being viewed by others


Introduction
In 1964, Lejeune et al1 described a proband with partial monosomy of a G group chromosome that was presumed to be 21. They described this as the ‘contretype’ (countertype) of trisomy 21. No non-mosaic complete monosomy of chromosome 21 or any other autosome has been confirmed in living individuals with modern techniques; however, segmental monosomy 21 has been reported. Published accounts indicate that chromosome 21 segmental monosomies are heterogeneous with regard to the severity of the phenotypes. In the most complete study to date Lyle et al2 reported 11 cases of partial monosomy 21 and outlined three affected regions with the associated phenotypic severity ranging from mild to severe to lethal (Figure 1a; see Discussion). In the present study we report on ten cases of partial monosomy 21 and we employ high density single nucleotide polymorphism (SNP) genotyping to further refine the monosomy breakpoints, and increase the number of patients described with clinical phenotypes associated with the three broad regions of monosomy.

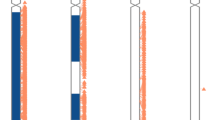
Genomic locations of chromosome 21 partial monosomies. Summary of chromosome 21 partial monosomy cases. (a) Three regions proposed by Lyle et al,2 separated by vertical dashed lines. (b) Physical positions of segmental monosomy in 11 cases reported by Lyle et al.2 Rectangles indicate the extent of the hemizygous deletion. (c) Positions of hemizygous deletions in three cases reported by Lindstrand et al.9 (d) Positions of hemizygous deletions in Cohort A. (e) Positions of hemizygous deletions in Cohort B. (f) Positions of hemizygous deletions in 12 DECIPHER database cases. (g) Ideogram of chromosome 21. All cases in panels b–f having a chromosomal abnormality (deletion, duplication, derivative chromosome, or translocation) on a chromosome other than 21, based on SNP, array CGH and/or karyotype data, are indicated with light grey rectangles. For full karyotypes and/or array data corresponding to panel b see Lyle et al2; for panels d, e see Supplementary File 3; for panel f see Supplementary File 8. Cases having only chromosome 21 segmental monsomy (without other identified chromosomal abnormalities) are indicated with black rectangles. All rectangles correspond to regions of deletions.
Materials and methods
Sample selection and SNP genotyping
Cohort A (Figure 1d) included three individuals seen at the Kennedy Krieger Institute and Johns Hopkins University having segmental chromosome 21 monosomies initially identified by G-banded karyotype. Additionally, karyotypes of their parents were performed to establish whether aberrations were de novo or inherited. All studies were performed with approval of the Johns Hopkins Institutional Review Board, and informed consent was obtained from the individuals involved. The karyotypes are listed in Table 1.
Patient sample genotyping was performed using the Illumina HumanHap550 v1.0 (Illumina Inc., San Diego, CA, USA) and Illumina HumanHap550 v3.0 Genotyping Beadchips. SNP genotypes were detected by whole-genome amplification and single-base extension per the Infinium II assay. Genotyping was performed at the Center for Inherited Disease Research (CIDR) at Johns Hopkins.
Cohort B (Figure 1e) consisted of seven individuals with cytogenetically identified chromosome 21 segmental monosomy from whom cell lines were available from the Coriell Genetic Cell Repository. Karyotypes are listed in Table 2. SNP data from the Affymetrix SNP 6.0 genotyping platform were generated at Coriell.
Chromosome 21 deletion breakpoint regions were defined using BeadStudio software (Illumina, Inc., San Diego, CA, USA), Genotyping Console software (Affymetrix, Inc., Santa Clara, CA, USA), custom scripts, and by manual inspection of SNP data. The last normal SNP before the deletion, the first and last SNP within the deletion, and the first normal SNP after the deletion were determined. Two main criteria were required to define a region as deleted: a reduced Log R Ratio, and a loss of heterozygosity as measured by a loss of SNPs clustered around 0.5 on the calculated B Allele Frequency.
Information about 12 additional cases of 21q partial monosomy was obtained from the DECIPHER database.3
Clinical reports
The clinical phenotypes for Cohort A are summarized in Table 1, and detailed descriptions are provided in Supplementary File 1. For Cohort B phenotypic data are summarized in Table 2 and in detail in Supplementary File 2. Phenotypic data were limited to those reported to Coriell by the case contributors.
Results
We determined the breakpoint regions for aberrations present in the ten cases of Cohorts A and B (Supplementary File 3). The regions of hemizygous deletions on chromosome 21 are summarized in Figure 1.
Cohort A
Comparison of parental genotype data to Patient 1 confirmed a de novo 21q terminal deletion (Supplementary Files 4 and 5), and allowed a refinement of the breakpoint (Supplementary File 3). The minimum deletion size was determined to be 4.86 Mb (chromosome 21 coordinates 42 047 505–46 909 417). Based on analysis with SNPtrio software (http://pevsnerlab.kennedykrieger.org),4 the deleted region exhibited a paternal SNP inheritance pattern. This indicated the deletion occurred on the maternally derived chromosome (Supplementary File 5). A total of 78 RefSeq annotated genes (Build 35) were included in the deleted region (Supplementary File 6). There was an additional, small hemizygous deletion on chromosome 20 that was also deleted in the mother (Supplementary File 3). While deletions in this region were reported to occur in apparently normal individuals in the Database of Genomic Variants (DGV), such deletions were absent from the DECIPHER database. Thus it is likely to be benign.
Analysis of the genotype data for Patient 2 confirmed a de novo 21q terminal hemizygous deletion (Supplementary Files 4 and 5) that completely overlapped the region deleted in Patient 1, but was larger in size with a minimum size of 5.68 Mb (chr21:41 229 212–46 909 417). Patient 1 was described as having the larger deletion by karyotype analysis, but the SNP genotyping data showed that patient 2 had the larger terminal deletion by 812–819 Kb. This offset contained seven RefSeq annotated genes (Supplementary File 6). In contrast to Patient 1, the SNP inheritance pattern indicated the deletion occurred on the paternally derived chromosome (Supplementary File 5).
For Patient 3, an interstitial 21q deletion was confirmed to be de novo after examination of the parental SNP data. The deletion included a minimum of 16.4 Mb of DNA in chromosomal bands 21q21.1–q22.11 (chr21:17 108 535–33 526 427) (Supplementary Files 3 and 4). Analyzing the SNP data for parent of origin identified the deletion on the paternal copy due to a maternal inheritance pattern in the deletion region (Supplementary File 5). The deletion included a total of 69 RefSeq annotated genes listed in Supplementary File 7, as well as microRNA hsa-miR155. We identified additional variants of ≈150 kb on chromosomes 4 (a hemizygous deletion affecting no genes, also deleted in the mother) and 14 (an amplification affecting two RefSeq genes, also present in an extra copy in the father; Supplementary File 3). For both of these additional variants there are overlaps with deleted or duplicated regions from the DGV. There were also several DECIPHER cases having overlapping abnormalities on chromosomes 4 and 14, but only involving larger affected regions and/or multiple chromosomal abnormalities. Thus, these additional variants are unlikely to contribute to the clinical phenotype.
Interstitial hemizygous deletion cases in Cohort B
Three cases obtained from Coriell had interstitial 21q deletions (Supplementary File 3). (1) For proband GM00137 we detected an interstitial hemizygous deletion of 15–18 Mb in chromosome 21q near the centromere. We also observed a 10.9 Mb deletion at the telomeric region of chromosome 4p to 4p16.1. (2) For case GM08210, we detected an interstitial 4.1 Mb hemizygous deletion in chromosome 21q near the centromere. There was also a hemizygous deletion of almost 4 Mb on 4p at the telomere. (3) We detected an interstitial hemizygous deletion in chromosome 21q near the centromere for proband GM06918, without prominent gains or losses on other chromosomes.
Terminal hemizygous deletion cases in Cohort B
Four cases had hemizygous terminal deletions (Supplementary File 3). (1) Analysis of SNP data for proband GM00983 indicated a terminal hemizygous deletion of about 8.6 Mb on 21q and a gain of 30.7 Mb on 10q25.1 to 10qter. (2) For case GM06136, we observed a terminal hemizygous deletion on 21q and a gain of material on the majority of 10p extending from near the centromere to the telomere (spanning 33 Mb). Cell line GM06136 had an apparent terminal deletion with the proximal breakpoint near the breakpoint for Cohort A Patient 2. The difference in their putative ends was less than 5 kb. (3) For proband GM09868, we observed a terminal hemizygous deletion on 21q involving 11.2 Mb. (4) We observed a terminal hemizygous deletion on 21q in GM19999 spanning 10.8 Mb.
In addition to the cases in Cohorts A and B, we identified 12 cases of hemizygous 21q deletion in the DECIPHER database.3 We plotted these (Figure 1e) and summarized the breakpoints (Supplementary File 8).
Discussion
Partial chromosome 21 monosomy is sometimes associated with severe clinical phenotypes, such as brain dysgenesis, extensive dysmorphic features, and heart defects.5, 6, 7, 8, 9 However, other patients present with very few dysmorphic features or congenital anomalies.10, 11 It is important to understand the genotype–phenotype correlation to elucidate the consequence of particular hemizygous deletions. In the present study we have characterized ten 21q monosomy cases, as well as described 12 cases from the DECIPHER database. We characterized the deletion breakpoint regions using high-density SNP arrays, and we provide descriptions of clinical phenotypes of varying severity.
We interpret the significance of our findings in the context of a model recently presented by Lyle et al2 and we extend that model. They reported 11 partial monosomy 21 cases (summarized in Figure 1b) as well as cases of partial trisomy 21, and they described three broad classes of partial 21 monosomy (Figure 1a). In the first region, deletions from the centromere to ∼31.2 Mb (spanning about 50 genes) are associated with a severe phenotype. In our study this region included Cohort A Patient 3, three Cohort B cases, and two DECIPHER cases (Figure 1d–f). Consistent with Lyle et al,2 deletions in this region tended to be large (three extended over 15 Mb). Although Patient 3 had a deletion encompassing approximately half of the q-arm of chromosome 21, she had few dysmorphic features and no detectable congenital brain, heart or kidney abnormalities. We note that two of the four cases in region 1 from Lyle et al2 as well as two of our cases had deletions or translocations involving chromosomes other than 21, and this may contribute to the clinical severity. Lindstrand et al9 reported three partial 21q deletion cases. Their patient 1, who died at the age of 5 months, had a complex intrachromosomal rearrangement involving four duplicated and four deleted segments, including a large deletion in Region 1. Their patient 3 had a 14 Mb hemizygous deletion in Region 1. This child had normal to late speech development, some problems in gross and fine motor functions, and normal to mildly delayed social, emotional, and cognitive development.
In the second region, from 31.2 to 36 Mb, Lyle et al2 found no cases that spanned the interval and only one case had a partial deletion. They suggested that deletions of this region, harboring ∼80 genes, produce a severe phenotype not compatible with survival. We report one Cohort A and three Cohort B cases having deletions overlapping Region 2. Lindstrand et al9 reported Patient 2 (Figure 1c) having an 11.9 Mb hemizygous deletion that spanned part of Region 1 and half of Region 2, with severely delayed psychomotor function and some dysmorphic features. Three DECIPHER cases occur in this region, two of which almost span it. This suggests that region 2 could be merged with Region 1. The severity of clinical presentation for deletions spanning this region was variable, and appears comparable with that of Region 1.
In agreement with Lyle et al,2 we also found hemizygous deletions in a third region, from ∼36–37.5 Mb to the telomere, to be the most common abnormality (two from Cohort A, four from Cohort B, eight from DECIPHER, seven from Lyle et al). Also in agreement with Lyle et al, the phenotypes were relatively mild. Although deletions in this region were common, the breakpoint locations were variable.
We depict the chromosome 21q deletion breakpoints for 36 cases in Figure 1. Of these, 16 cases involve no other identified chromosomal abnormalities, whereas 20 cases involve aneuploidy and/or a translocation on a chromosome other than 21. We may reasonably exclude these latter cases from the analysis of genotype/phenotype correlations because the effects of additional chromosomal abnormalities are too complex to interpret. Our interpretation of the 16 cases as a group still suggests that Regions 1 and 2 may be combined. There is still considerable variability in the severity of the phenotype considering this larger region. This interpretation is consistent with that of Lindstrand et al9 who reviewed 35 published cases of 21q deletion. They proposed that a small region (from 33.83 to 33.99 Mb), including the ITSN1 gene, is deleted in most patients with mental retardation or severe developmental delay; and further they suggested that the neighboring genes KCNE1, RCAN1, CLIC6, and RUNX1 form a critical region of severe cardiac malformations. This region was not affected in cases from Cohort A or B, and although case 42 from Lyle et al does include this region (Figure 1b), that patient also has a X chromosome aneuploidy that could impact the cardiac phenotype. This highlights the current challenge of identifying genes responsible for developmental abnormalities in partial 21q deletion cases.
We conclude that the variability of clinical phenotypes reflect the variable sizes of the hemizygous deletion, the location of the deletion, the influence of variants at other chromosomal loci in each individual's genome, and other environmental factors. As we continue to determine breakpoint locations and standardize the way clinical phenotype data are collected, we may further discern candidate loci for particular phenotypes associated with these deletions.
References
Lejeune J, Berger R, Rethore MO et al: Partial monosomy for a small acrocentric chromosome. C R Hebd Seances Acad Sci 1964; 259: 4187–4190.
Lyle R, Béna F, Gagos S et al: Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur J Hum Genet 2009; 17: 454–466.
Firth HV, Richards SM, Bevan AP et al: DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 2009; 84: 524–533.
Ting JC, Roberson EDO, Miller ND et al: (2007) Visualization of uniparental inheritance, Mendelian inconsistencies, deletions, and parent of origin effects in single nucleotide polymorphism trio data with SNPtrio. Hum Mutation 2007; 28: 1225–1235.
Yao G, Chen X-N, Flores-Sarnet L et al: Deletion of chromosome 21 disturbs human brain morphogenesis. Genetics in Medicine 2006; 8: 1–7.
Keren B, Bernardin C, Toutain A et al: Pure proximal deletion of chromosome 21 and kyphosis. Eur J Med Genet 2007; 50: 469–474.
Takhar J, Malla AK, Siu V et al: An interstitial deletion of the long arm of chromosome 21 in a case of a first episode of psychosis. Acta Psychiatrica Scandinavica 2002; 106: 71–74.
Chettouh Z, Croquette M-F, Delobel B et al: Molecular mapping of 21 features associated with partial monosomy 21: involvement of the APP-SOD1 region. Am J Hum Genet 1995; 57: 62–71.
Lindstrand A, Malmgren H, Sahlén S et al: Detailed molecular and clinical characterization of three patients with 21q deletions. Clin Genet 2010; 77: 145–154.
Ehling D, Kennerknecht I, Junge A et al: Mild phenotype in two unrelated patients with a partial deletion of 21q22.2-q22.3 defined by FISH and molecular studies. Am J Med Genet Part A 2004; 131A: 265–272.
Tinkel-Vernon H, Finkernagel S, Desposito F et al: Patient with a deletion of chromosome 21q and minimal phenotype. Am J Med Genet Part A 2003; 120A: 142–143.
Acknowledgements
The authors sincerely thank the families participating in this study. We thank Nathaniel D. Miller for helpful comments on the manuscript. This work was supported in part by NIH Grant HD024061 (J.P. and GHT) and the Stem Cell Resource Center at Johns Hopkins Institute of Cell Engineering (JP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Roberson, E., Wohler, E., Hoover-Fong, J. et al. Genomic analysis of partial 21q monosomies with variable phenotypes. Eur J Hum Genet 19, 235–238 (2011). https://doi.org/10.1038/ejhg.2010.150
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.150
Keywords
This article is cited by
-
De novo mosaic and partial monosomy of chromosome 21 in a case with superior vena cava duplication
Molecular Cytogenetics (2020)
-
Monosomy chromosome 21 compensated by 21q22.11q22.3 duplication in a case with small size and minor anomalies
Molecular Cytogenetics (2018)
-
Proximal 21q deletion as a result of a de novo unbalanced t(12;21) translocation in a patient with dysmorphic features, hepatomegaly, thick myocardium and delayed psychomotor development
Molecular Cytogenetics (2016)
-
Deletion 21q22.3 and duplication 7q35q36.3 in a Colombian girl: a case report
Journal of Medical Case Reports (2016)
-
Dissection of partial 21q monosomy in different phenotypes: clinical and molecular characterization of five cases and review of the literature
Molecular Cytogenetics (2016)

{kind=link}
{kind=link}