
Abstract
Studies of large sets of single nucleotide polymorphism (SNP) data have proven to be a powerful tool in the analysis of the genetic structure of human populations. In this work, we analyze genotyping data for 2841 SNPs in 12 sub-Saharan African populations, including a previously unsampled region of southeastern Africa (Mozambique). We show that robust results in a world-wide perspective can be obtained when analyzing only 1000 SNPs. Our main results both confirm the results of previous studies, and show new and interesting features in sub-Saharan African genetic complexity. There is a strong differentiation of Nilo-Saharans, much beyond what would be expected by geography. Hunter-gatherer populations (Khoisan and Pygmies) show a clear distinctiveness with very intrinsic Pygmy (and not only Khoisan) genetic features. Populations of the West Africa present an unexpected similarity among them, possibly the result of a population expansion. Finally, we find a strong differentiation of the southeastern Bantu population from Mozambique, which suggests an assimilation of a pre-Bantu substrate by Bantu speakers in the region.
Similar content being viewed by others

Introduction
Human population genetics has recently completed a circle. It started with the so-called classical polymorphisms (ie, blood groups and other protein polymorphisms) that were analyzed on the basis of their allele frequencies with potent statistical instruments such as principal component analysis (PCA). This era culminated with the landmark publication of the magnificent book by Cavalli-Sforza et al1; however, a few shortcomings of classical polymorphisms can be pointed out: relatively few loci were used; their relationship to the underlying genetic variation was mostly unknown; and they could have been subjected to confounding by natural selection. PCR and automated sequencing heralded the uniparental marker era: mtDNA and the non-recombining region of the Y-chromosome could be routinely analyzed, and a firm phylogeography could be established for both genomic regions, allowing the dissection of population structure with unprecedented precision and reliability. But yet, they behave as just two loci, wherein natural selection cannot be ruled out, as well as the peculiarities associated with their sex-specific transmission. Another technological development spearheaded a new breakthrough in human population genetics: single nucleotide polymorphism (SNP) array genotyping platforms have made it affordable to genotype hundreds of thousands of markers. The results are again treated in terms of allele frequencies and subjected to PCA or to newer techniques, such as Bayesian classification algorithms. Now, the whole genome is covered, and the action of selection is masked by a vast majority of putatively neutral markers.
The genetics of African populations, of paramount interest given the recent African origin of humankind, has been through the full cycle of studies. Cavalli-Sforza et al1 identified a north–south gradient in the continent that could be attributed to the Bantu expansion, whereas other principal components had a less clear interpretation. African mtDNA phylogeography was firmly established by Salas et al2, who described the structure of maternal lineages in the continent, and identified some haplogroups involved in human expansions such as the Bantu expansion. Deep analyses of maternal lineages in African hunter gatherers (Khoisan speakers and Pygmies) have revealed a clearly structured phylogeny for the mtDNA.3, 4, 5 A number of papers have approached both the general and the more local aspects of non-recombining part of the Y-chromosome in Africa,6, 7 although its phylogeographic structure has not been as refined as its maternal counterpart. More recently, in a landmark paper by Tishkoff et al8, 1327 nuclear microsatellite markers were analyzed in 121 African populations, identifying a number of layers in the African population structure that could be related to history, language, and geography. The continent, south of the Sahara, seems to be dominated by a component mostly correlated with Niger-Congo speaking populations, whereas other components are found in the Sahel, among Nilo-Saharan speakers, and in Afro-Asiatic speakers in the north and northeast. Among the hunter-gatherer populations, the Khoisan-speaking Hadza of Tanzania were clearly distinct, whereas Pygmies could not be discriminated from the South African Khoisan. At higher discrimination level, western Pygmies became distinct, but the eastern Pygmies remained similar to the Khoisan. In another recent publication, Bryc et al 9 analyzed SNP data obtained from West Africans (and African Americans), revealing a structure reflecting primarily language and secondarily geographical distances. Unfortunately, this work is restricted mainly to populations in Central West Africa, around the Gulf of Guinea. Both of these recent studies have greatly advanced our understanding of the genetic structure of Sub-Saharan Africa. However, both in Tishkoff et al and Bryc et al as well as in previous works, the area between Central and South Africa remains under sampled. Clearly, southeast Africa is a key geographical zone to understand the Bantu expansion routes, making it a region of particular interest for the population history of sub-Saharan Africa.
Data
In a case-control study for placental malaria, we obtained, with appropriate informed consent, 180 cases and 180 controls from Mozambique, in southeast Africa. These samples were genotyped with the Affymetrix GeneChip Human Immune and Inflammation 9K SNP Kit (Santa Clara, CA, USA), resulting in a total of 279 samples with reliable data after stringent quality control. Other African samples with genome wide SNP data available are Biaka Pygmies, Mbuti Pygmies, Mandenka, Yoruba, San, and Bantu-speakers from the HGDP panel10, and the Maasai, Luhya, Yoruba, and African-Americans from HapMap Phase 3 (http://hapmap.ncbi.nlm.nih.gov/). The intersection of all arrays provides a common set of 2841 SNPs with genotype data for all populations (see Supplementary Information for details).
Results
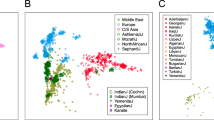
First, we wanted to test whether the number of SNPs available (2841 SNPs) provides enough genetic resolution to detect any structure in African populations and provide a reference for the number of SNPs needed in population studies. To that effect, we combined the global Human Genome Diversity Panel (HGDP) and HapMap phase 3 genotype data (∼460 000 SNPs) and subjected them to PCA (see Supplementary Information for details). Results are similar to those obtained with the HGDP samples, with the first and second PC (Figure 1a) separating East Asia (upper left corner of the plot) from Europe (bottom centre) and sub-Saharan Africa (upper right).10, 11 The same structure is recovered when random subsamples of 100 000 (Figure 1b), 10 000 (Figure 1c), and 1000 (Figure 1d) SNPs are considered, although inter-individual variation increases. A random set of 2841 SNPs from this pooled HGDP-HapMap dataset (Figure 1e) performs similarly to the set of 2841 SNPs related to immunity and inflammation (Figure 1f), despite of the slightly reduced interpopulation differentiation of the latter, which is expected as they are gene-based SNPs.12 We can conclude that the common set of 2841 SNPs genotyped is an appropriate tool to study population structure in African populations; in general, worldwide patterns are evident and robust when using a minimum of 1000 SNPs.

PCA of merged HGDP and Hap Map 3 samples. Panels show the results of the PCA for the full merged set of SNPs (460 147 SNPs) (a), for random subsets of 100 000 (b), 10 000 (c), 1000 (d), and 2841 SNPs (e), as well as for the 2841 SNPs in the merged analysis set including the samples from Mozambique (f). As can be seen, the general pattern of differentiation is reproduced even using only a random subset of 1000 SNPs. Different colors indicate continental region for the respective populations . Abbreviations: AME, Americas; CSASIA, Central and South Asia; EASIA, East Asia; EUR, Europe; MENA, Middle East and North Africa; OCE, Oceania; SSAFR, sub-Saharan Africa.
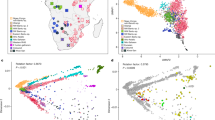
Next, we applied PCA13 and STRUCTURE14, 15 to 775 individuals in 11 populations of sub-Saharan African descent. The first PC (Figure 2a) and STRUCTURE with K=2 (Figure 3) separate the Nilo-Saharan-speaking Maasai from all other populations, with neighboring Luhya and African Americans in an intermediate position. Both the second PC and K=3 separate the hunter-gatherer samples, presumably ancestral Pygmy and San populations from the rest. The third PC allows us to discriminate between western/central (Mandenka, Yoruba), eastern (Maasai, Luhya), and southeastern populations (Mozambique), irrespectively of language family. This is the PC that is mostly correlated with geography (Figure 2c), and the fact that it is the third rather than the first component, as would be expected if isolation by distance was the predominant force shaping genetic diversity,16 implies that directional population movements (such as the Bantu expansion) and barriers to gene flow (such as that between food producers and hunter gatherers) are more relevant than geographic distance to understand the genetic landscape of sub-Saharan Africa. The distinction between west and southeast Africa is also shown with K=4; at K=5, the Niger-Congo speaking Luhya are separated from the rest. The new component that appears at K=6 is restricted to African Americans and Biaka Pygmies, and is the last component that can be attributed to specific populations.

PCA of sub-Saharan African populations. Panels show plots of the first three principal components obtained from the 11 sub-Saharan African populations. (a) First and second components. (b) First and third components. (c) Biplot of rotated PC1 and PC3 superimposed onto a map of Africa. Geographical locations of the populations are indicated by their names and their respective enlarged plot symbols. Different colors indicate linguistic or cultural group for the respective populations (green: Nilo-Saharan; blue: Niger-Congo; orange: Hunter Gatherer; grey: admixed).

STRUCTURE results for sub-Saharan African populations. Depicted are the results of five runs each for the number of clusters ranging from K=2 to K=7, combined using CLUMPP.
Discussion
The preceding results are in agreement with what was found previously by Tishkoff et al using microsatellites, and goes beyond with new findings and refinement of previous genetic studies:
-
i)
The main distinction is among Niger-Congo groups and the rest, including Nilo-Saharan speakers and hunter gatherers (with the Khoisan having preserved their ancestral language but not Pygmies). Among Niger-Congo populations, geography is the main factor explaining the genetic differences, with a remarkable similarity among western populations (Yorubas and Mandenka), which could reflect a burst in the expansion to the west, related to iron technology and Niger-Congo languages.
-
ii)
The southeastern Bantu from Mozambique are remarkably differentiated from the western Niger-Congo speaking populations, such as the Mandenka and the Yoruba, and also differentiated from geographically closer Eastern Bantu samples, such as Luhya. These results suggest that the Bantu expansion of languages, which started ∼5000 years ago at the present day border region of Nigeria and Cameroon, and was probably related to the spread of agriculture and the emergence of iron technology,17, 18, 19 was not a demographic homogeneous migration with population replacement in the southernmost part of the continent, but acquired more divergence, likely because of the integration of pre-Bantu people. The complexity of the expansion of Bantu languages to the south (with an eastern and a western route20), might have produced differential degrees of assimilation of previous populations of hunter gatherers. This assimilation has been detected through uniparental markers because of the genetic comparison of nowadays hunter gatherers (Pygmies and Khoisan) with Bantu speaker agriculturalists.2, 21, 22, 23, 24 Nonetheless, the singularity of the southeastern population of Mozambique (poorly related to present Khoisan) could be attributed to a complete assimilation of ancient genetically differentiated populations (presently unknown) by Bantu speakers in southeastern Africa, without leaving any pre-Bantu population in the area to compare with.
-
iii)
The difference between hunter-gatherers and the rest of South Saharan populations is important but it is not the main trait in the African genetics. To note is the strong similarity among the three studied populations, with no specific Pygmy component, but an important Bantu introgression (as seen in K=3) in Biaka Pygmies. Pygmies should be included along with Khoisan in the search for deep-rooted African and Human lineages. Moreover, the specific component that identifies the three hunter-gatherer populations is found at a small amount in all other African populations, as a possible result of introgression with previous settlers of most African territory.
As a more general observation, we found that as little as 1000 genome wide SNPs are enough to robustly recover the patterns of genetic structure among worldwide populations. It has to be noted that even though this low number seems to be sufficient for inferring major demographic events and broad population structure, it remains doubtful whether it will be sufficient for more fine-scale inference, as for example within a genetically uniform region such as Europe. Nevertheless, the high level of genetic structure in sub-Saharan Africa allows us to be confident in our conclusions. Furthermore, the fact that our dataset of 2841 SNPs has only limited fine-scale resolution makes the observed strong differentiation of the population from Mozambique even more striking. The genetic analysis of a large number of SNPs is thus providing a robust tool to refine our understanding of past populations history.
References
Cavalli-Sforza LL, Menozzi P, Piazza A : The History and Geography of Human Genes. Princeton University Press: Princeton, 1994.
Salas A, Richards M, De la Fe T et al: The making of the African mtDNA landscape. Am J Hum Genet 2002; 71: 1082–1111.
Batini C, Coia V, Battaggia C et al: Phylogeography of the human mitochondrial L1c haplogroup: genetic signatures of the prehistory of Central Africa. Mol Phylogenet Evol 2007; 43: 635–644.
Quintana-Murci L, Quach H, Harmant C et al: Maternal traces of deep common ancestry and asymmetric gene flow between Pygmy hunter-gatherers and Bantu-speaking farmers. Proc Natl Acad Sci USA 2008; 105: 1596–1601.
Behar DM, Villems R, Soodyall H et al: The dawn of human matrilineal diversity. Am J Hum Genet 2008; 82: 1130–1140.
Cruciani F, Santolamazza P, Shen P et al: A back migration from Asia to sub-Saharan Africa is supported by high-resolution analysis of human Y-chromosome haplotypes. Am J Hum Genet 2002; 70: 1197–1214.
Berniell-Lee G, Calafell F, Bosch E et al: Genetic and demographic implications of the Bantu expansion: insights from human paternal lineages. Mol Biol Evol 2009; 26: 1581–1589.
Tishkoff SA, Reed FA, Friedlaender FR et al: The genetic structure and history of Africans and African Americans. Science 2009; 324: 1035–1044.
Bryc K, Auton A, Nelson MR et al: Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc Natl Acad Sci USA 2010; 107: 786–791.
Li JZ, Absher DM, Tang H et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100–1104.
Jakobsson M, Scholz SW, Scheet P et al: Genotype, haplotype and copy-number variation in worldwide human populations. Nature 2008; 451: 998–1003.
Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L : Natural selection has driven population differentiation in modern humans. Nat Genet 2008; 40: 340–345.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Pritchard JK, Stephens M, Donnelly P : Inference of population structure using multilocus genotype data. Genetics 2000; 155: 945–959.
Falush D, Stephens M, Pritchard JK : Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 2003; 164: 1567–1587.
Novembre J, Stephens M : Interpreting principal component analyses of spatial population genetic variation. Nat Genet 2008; 40: 646–649.
Vansina J : New Linguistic evidence and ‘the bantu expansion’. J African Hist 1995; 36: 173–195.
Phillipson DW : African Archaeology. Cambridge University Press: Cambridge, 1993.
Newman JL : The Peopling of Africa: A Geographic Interpretation. Yale University Press: New Haven, London, 1995.
Oslisly R : The middle Ogooué valley, Gabon: cultural changes and palaeoclimatic implications of the last fourmillenia. Azania 1995; 39–40: 324–331.
Pereira L, Gusmao L, Alves C, Amorim A, Prata MJ : Bantu and European Y-lineages in sub-Saharan Africa. Ann Hum Genet 2002; 66: 369–378.
Pereira L, Macaulay V, Torroni A, Scozzari R, Prata MJ, Amorim A : Prehistoric and historic traces in the mtDNA of Mozambique: insights into the Bantu expansions and the slave trade. Ann Hum Genet 2001; 65: 439–458.
Plaza S, Salas A, Calafell F et al: Insights into the western Bantu dispersal: mtDNA lineage analysis in Angola. Hum Genet 2004; 115: 439–447.
Beleza S, Gusmao L, Amorim A, Carracedo A, Salas A : The genetic legacy of western Bantu migrations. Hum Genet 2005; 117: 366–375.
Acknowledgements
This research was supported by the Dirección General de Investigación, Ministerio de Ciencia y Tecnología, Spain (grants SAF2007-63171, BFU2007-63657) and Direcció General de Recerca, Generalitat de Catalunya (2009 SGR 1101). SNP genotyping services were provided by the Spanish ‘Centro Nacional de Genotipado’ (CEGEN; http://www.cegen.org); Bioinformatic services were kindly provided by the Genomic Diversity node, Spanish Bioinformatic Institute (http://www.inab.org). MS was supported by a PhD fellowship from the Programa de becas FPU del Ministerio de Educación y Ciencia, Spain (AP2005-3982).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Sikora, M., Laayouni, H., Calafell, F. et al. A genomic analysis identifies a novel component in the genetic structure of sub-Saharan African populations. Eur J Hum Genet 19, 84–88 (2011). https://doi.org/10.1038/ejhg.2010.141
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.141
Keywords
This article is cited by
-
The influence of habitats on female mobility in Central and Western Africa inferred from human mitochondrial variation
BMC Evolutionary Biology (2013)
-
Prediction of people’s origin from degraded DNA—presentation of SNP assays and calculation of probability
International Journal of Legal Medicine (2013)
