
Abstract
Severe congenital neutropenia type 4 (SCN4) is an autosomal recessive condition, which was defined recently with identification of the causative mutations in G6PC3. To date there are only three reports in the literature describing patients with SCN4 with mutations in the G6PC3 gene. We report four individuals with SCN4 who belong to a single large consanguineous kindred. We provide an overview of the non-haematological features of the condition with a focus on the adult phenotype, which has not been previously described in detail. We show that the superficial venous changes seen in SCN4 patients can develop into varicose veins and venous ulcers in adulthood. We review the range of congenital anomalies associated with SCN4. We demonstrate that secundum atrial septal defect, patent ductus arteriosus and valvular defects are the most frequent cardiac anomalies in SCN4. Drawing parallels with type 1 glycogen storage disease, we propose that poor growth of prenatal onset, mild-to-moderate learning disability, primary pulmonary hypertension, delayed or incomplete puberty, hypothyroidism and dysmorphism likely represent features of this syndrome. We also suggest monitoring for lipid anomalies, and kidney and liver function in affected patients. Delineation of the SCN4 phenotype may help in appropriate treatment and management and provide further insights into the pathogenesis of this multisystem disease.
Similar content being viewed by others
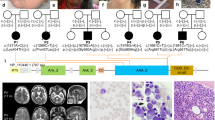
Introduction
The congenital neutropenias are a group of heterogeneous disorders. To date, 17 causative genes have been identified for syndromic and non-syndromic causes.1 Severe congenital neutropenia type 4 (SCN4, OMIM 612541) was recently defined with identification of the causative mutations in G6PC3 (Gene ID 92579) and to date there are only three reports in the literature describing 15 patients in total.2, 3, 4 SCN4 has a distinct phenotype showing the presence of a prominent superficial venous pattern and an increased susceptibility to infection. Other described features include intermittent thrombocytopenia, hepatosplenomegaly, congenital heart defects, urogenital malformations, cleft palate, inguinal hernia, microcephaly, sensorineural hearing loss and myopathy.3
We report four individuals from a single large consanguineous Israeli kindred of Arab-Muslim origin with SCN, a prominent superficial venous pattern and additional clinical features. The pedigree is shown in Figure 1 and the clinical details are provided below.

Pedigrees of one large and one small nuclear family are represented. Both the families belong to a single kindred although the exact relationship between them is not certain.
Clinical description
Patient 1 (V-7) is a 29-year-old female born to first cousin parents. She was small for gestation at birth. Her adult height is on the second centile and her weight and occipito-frontal circumference (OFC) are on the fifth centile. She also has mild learning disability and required special education. She has irregular menses. She has suffered from recurrent pneumonia, otitis media and perianal abscesses. Significant results of her haematological and biochemical investigations are summarized in Table 1. On examination, she has a prominent superficial venous pattern and varicose veins. She also shows mild kyphosis, tapering fingers and clinodactyly of the fourth and fifth fingers.
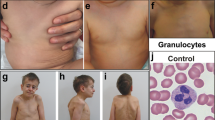
Patient 2 (V-13) is a 26-year-old man. He is V-7's second cousin on the maternal side and third cousin on the paternal side. At birth he was small for dates and failed to thrive in infancy. His current height is below the 0.4th centile and weight is on the second centile. Agenesis of the left kidney and right hydronephrosis were noted in childhood. His puberty was delayed. He also has mild learning disability and went to a special school. He has experienced recurrent episodes of pneumonia and otitis media. His relevant haematological and biochemical investigation results are presented in Table 1. On examination he has a prominent superficial venous pattern and varicose veins, which have resulted in a venous stasis ulcer. He has poorly developed secondary sexual characteristics and gynaecomastia. He has full cheeks (Figure 2a) and fat deposition around his trunk. His anterior and posterior hairlines are low (Figure 2b) and he has broad thumbs.

(a, b) Front and profile of patient 2 showing full cheeks (similar to what is seen in GSD1), prominent lips and low hairlines.
One of his younger brothers (V-10) died soon after birth because of a congenital heart defect, about which more details are not available. He has an older sister (V-14) who has learning difficulties but does not share any other clinical features of SCN4.
Patient 3 (V-17) was V-13's double first cousin. Patent ductus arteriosus (PDA) and secundum atrial septal defect (ASD) were noted at birth. Her adult height, weight and OFC were all on the second centile. Menarche was not delayed but secondary sexual characteristics were slow to develop. She also had mild learning difficulties and went to a special-needs school. She suffered from recurrent urinary tract infections and herpes stomatitis. She died at the age of 25 years because of sepsis. Her peripheral blood counts and bone-marrow aspiration results are summarized in Table 1. On examination she had prominent and widespread superficial venous pattern and varicose veins on the lower legs. She had acne on her face and trunk. Her breasts and pubic hair were between Tanner stages 3 and 4 in adulthood. She had bilateral keratoconus.
Patient 4 (V-21) is a 2-year-old boy born to first cousin parents. His parents belong to the same kindred of the first three patients described above, although the exact relationship is not known. He was diagnosed prenatally to have pulmonary valve stenosis. At 8 days of age he underwent pulmonic balloon valvuloplasty. PDA and secundum ASD were also noted. Soon after birth he developed pulmonary hypertension of unknown cause, which was treated with sildenafil until 8 months of age. He had bilateral undescended testis and gastro-oesophageal reflux. He was also found to have raised gamma-glutamyl transferase and calcifications in the liver at 7 days of age. At age 3 months he was diagnosed with choledocholithiasis and was treated with ursodeoxycholic acid. He is still failing to thrive and has global mild-to-moderate developmental delay. His current length is on the fifth centile, weight below 0.4th centile and OFC on the 0.4th centile. He suffers with recurrent otitis media. His pretreatment peripheral blood counts and bone-marrow aspiration results (Supplementary Figure 1) are provided in Table 1. On examination he has plagiocephaly, sparse scalp hair, mid-face hypoplasia and a tented mouth. He also has a prominent superficial venous pattern on his head, a pectus carinatum and a small umbilical hernia. He has loose skin on the palms and soles and an uneven fat distribution.
Materials and methods
Ethical approval for this study was obtained from the University of Manchester (06138) and NHS ethics committee (06/Q1406/52). DNA was extracted from lymphocytes from the four affected individuals using standard protocol. All six exons of G6PC3 were amplified by polymerase chain reaction in the first three patients using the primers described by Boztug et al.3 In patient 4, only exon 6 of G6PC3 was amplified. V-14 has not been tested for the familial mutation. Genomic DNA sequence was analyzed using ABI 3130 (Applied Biosystems, Carlsbad, CA, USA) and Staden software package (http://staden.sourceforge.net).
Results
All four patients were homozygous for the c.758G>A mutation in G6PC3 (Figures 3a and b), which results in substitution of arginine to histidine at position 253 (p.R253H). This mutation has been described previously.3 We were unable to establish if this mutation was on a common haplotype with previously reported cases or whether this residue represents a mutational hotspot. Previous cases with the same mutation have been of Turkish-Armenian background, which would be consistent with a shared Mediterranean ancestry with the Israeli Arabs.5

(a) Homozygous mutation c.758G>A in exon 6 of G6PC3 of patient 1. (b) Corresponding normal sequence from a control subject.
Discussion
Glucose-6-phosphatase catalyzes the final step in glycogenolysis, that is, the conversion of glucose-6-phosphate to glucose. This step takes place in the endoplasmic reticulum (ER). Glucose-6-phosphatase is encoded by a gene-family consisting of G6PC1, G6PC2 and G6PC3.6 G6PC1 is expressed in the liver, kidney and small intestine. Homozygous loss-of-function mutations in G6PC1 cause type 1a glycogen storage disease (GSD1a).7 GSD1a is characterized by fasting hypoglycaemia, hepatomegaly, growth retardation, fasting lactic acidosis, hyperlipidemia and hyperuricemia. Long-term complications of GSD1a include hepatic adenomas, pancreatitis, gout, renal failure, pulmonary hypertension, polycystic ovaries, osteoporosis, platelet dysfunction and mild-to-moderate learning difficulties.8
The transport of glucose-6-phosphate from the cytosol to ER is facilitated by glucose-6-phosphate translocase, encoded by the ubiquitously expressed SLC37A4.9 Homozygous mutations in SLC37A4 result in GSD1b, which has a more severe phenotype than GSD1a and, of note, also causes chronic neutropenia.
G6PC3 is expressed ubiquitously and is important in maintaining neutrophil viability.3 Loss of G6PC3 function results in increased susceptibility to apoptosis in skin fibroblasts, neutrophils and myeloid cells. Increased apoptosis is likely because of decreased intra-cellular glucose concentrations and mediated through an ER stress mechanism and may predispose to associated congenital malformations.
Including the four patients presented here, a total of 19 SCN4 cases have now been described in the literature.2, 3, 4 Congenital neutropenia, frequent infections and prominent superficial veins are consistent features in these patients. We provide an overview of the non-haematological features of the condition with a focus on the adult phenotype, which has not previously been described in detail. We draw parallels between the phenotype of SCN4 patients with GSD1a because both G6PC3 and G6PC1 code for the enzyme glucose-6-phosphatase and with GSD1b because of the known functional relationship of the enzyme with glucose-6-phosphate transporter. A summary of major clinical features is provided in Table 2.
Venous anomalies
The prominent superficial venous pattern, which is the most distinctive non-haematological feature of SCN4, is less evident in infancy and gradually becomes more prominent with age. Considering the patients described here, it is clear that these vascular changes can develop into varicose veins in adulthood resulting in venous ulcers. Patients with SCN4 should be monitored for this complication.
Cardiac anomalies
Patients 3 and 4 were diagnosed with PDA and secundum ASD. Patient 4 also had PS. Combining our data with that presented by Boztug et al3 and Xia et al,4 13 out of 19 patients have at least one congenital cardiac malformation (see Table 2 for details). Secundum ASD and valvular defects are the most frequent cardiac anomalies in SCN4.
Pulmonary vasculature anomalies
One patient in the series presented by Boztug et al3 had pulmonary venous malformation. The congenital cardiac malformations (PS, ASD and PDA) observed in patient 4 do not normally result in pulmonary hypertension in early life. This leads us to conclude that the pulmonary disease in this patient was primary. Of note, Dursun et al10 recently reported a pair of Turkish siblings, one boy and one girl, with primary pulmonary hypertension, leucopenia and secundum ASD. Both the siblings with Dursun syndrome (OMIM 613034) demonstrated a phenotype that is strikingly similar to that of the children described with SCN4. Primary pulmonary hypertension is also a well-recognized feature of GSD111 and may be an additional feature of SCN4.
Growth
Poor growth of prenatal onset may also be a core feature of SCN4. All four affected individuals presented here showed intrauterine growth retardation and demonstrated a failure to thrive in infancy. Childhood stature and/or final adult height seem to be at the lower end of the normal range in all the four cases. The height and weight of the boy described by Arostegui et al2 were below the third percentile and at least 7 of the 12 patients reported by Boztug et al3 demonstrated growth retardation or height/weight at the lower end of the normal range. We note that female g6pc3−/− mice exhibit growth retardation,12 but in humans this features seems not to be sex limited.
Development
All four patients described here have mild-to-moderate developmental delay and/or learning disability. All three adult patients required special education and none have ever been in paid employment. Patient 2 has a sibling with learning disability who does not share the rest of the SCN4 phenotype and therefore the possibility of a different cause for learning difficulties in this kindred cannot be ruled out. The male reported by Arostegui et al2 also had learning difficulties, while no specific comments were made about neurodevelopmental aspects in the series described by Boztug et al3 and Xia et al.4 We suggest that the neurodevelopmental aspect of the SCN4 phenotype warrants further investigation.
Endocrine dysfunction
Of 15 previously reported patients, only 2 were beyond pubertal age – a 22-year-old male described by Arostegui et al2 and a 17-year-old male described by Boztug et al.3 The ages of the two patients described by Xia et al4 were not given. Three of the patients described here are over the age of 25 years, thus providing an insight into the adult SCN4 phenotype. Raised TSH was found in patients 1 and 2, suggesting that hypothyroidism may represent a feature of SCN4. Poor secondary sexual development was seen in two of our patients (patients 2 and 3) and we note that g6pc3−/− mice exhibit significant reduction in g6p hydrolytic activity in the testes, although without any known phenotypic consequence. We note also that delayed puberty is a known feature of GSD1. We suggest that evaluation of pubertal development in adolescents and thyroid function assessment should be considered in the follow-up of SCN4 affected patients.
Lipid abnormalities
Patients with GSD1 often have dyslipidemia, which is thought to be due to increased de novo lipogenesis and delayed conversion of very low-density lipoproteins to intermediate lipoprotein particles.13 Patient 1, the oldest in our series, had low HDL levels. She also had persistently increased amylase activity. The risk of pancreatitis because of dyslipidemia is increased in GSD1.14 Our findings suggest that lipid profile should be monitored in adults with SCN4.
Liver and kidney function
Findings of raised gamma-glutamyl transferase, liver calcifications and choledocholithiasis in patient 4, and elevated uric acid level in patient 1, indicates that liver function, kidney function and uric acid levels should be monitored in SCN4 similar to patients with GSD1.
Other congenital anomalies and minor dysmorphism
Inguinal hernia, cryptorchidism, genital dysplasia cleft palate and urachal fistula have already been described in patients with SCN4.3 To this list we can now add renal agenesis, hydronephrosis (patient 2) and umbilical hernia (patient 4). Skeletal anomalies such as pectus carinatum, clinodactyly, broad thumbs and kyphosis could also be a part of this syndrome.
Additional dysmorphic features can provide useful clues towards a specific diagnosis. Proper delineation of this aspect of the SCN4 phenotype will require a larger series of patients from different ethnic backgrounds. Considering the features observed in patient 4, it may be that mid-face hypoplasia, sparse scalp hair and loose skin represent features of SCN4. It is not clear if loose skin is an independent feature or secondary to failure to thrive. In adolescents and adults, the fullness of cheeks is reminiscent of GSD1, and a truncal fat distribution might provide added clues to the diagnosis.
Conclusion
SCN4 is a recently described autosomal recessive condition whose phenotypic extent and natural history is still incompletely defined. It is important to recognize this syndrome so that appropriate treatment and monitoring can be instigated. Further studies into larger cohorts of patients are needed to determine the extent of possible complications, clarify associated dysmorphic features and determine if developmental delay and learning difficulties are consistent features of the syndrome. It is clear that significant inter- and intra-familial variability can occur. Delineation of the phenotype will also provide further insights into the mechanisms of this disease.
References
Klein C : Molecular basis of congenital neutropenia. Haematologica 2009; 94: 1333–1336.
Arostegui JI, de Toledo JS, Pascal M, Garcia C, Yague J, Diaz de Heredia C : A novel G6PC3 homozygous 1-bp deletion as a cause of severe congenital neutropenia. Blood 2009; 114: 1718–1719.
Boztug K, Appaswamy G, Ashikov A et al: A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med 2009; 360: 32–43.
Xia J, Bolyard AA, Rodger E et al: Prevalence of mutations in ELANE, GFI1, HAX1, SBDS, WAS and G6PC3 in patients with severe congenital neutropenia. Br J Haematol 2009; 147: 535–542.
Arnaiz-Villena A, Karin M, Bendikuze N et al: HLA alleles and haplotypes in the Turkish population: relatedness to Kurds, Armenians and other Mediterraneans. Tissue Antigens 2001; 57: 308–317.
Hutton JC, O’Brien RM : Glucose-6-phosphatase catalytic subunit gene family. J Biol Chem 2009; 284: 29241–29245.
Lei K-J, Shelly LL, Pan C-J, Sidbury JB, Chou JY : Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a. Science 1993; 262: 580–583.
Janecke AR, Mayatepek E, Utermann G : Molecular genetics of type 1 glycogen storage disease. Mol Genet Metab 2001; 73: 117–125.
Veiga-da-Cunha M, Gerin I, Chen Y-T et al: The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type I non-a. Eur J Hum Genet 1999; 7: 717–723.
Dursun A, Ozgul RK, Soydas A et al: Familial pulmonary arterial hypertension, leucopenia, and atrial septal defect: a probable new familial syndrome with multisystem involvement. Clin Dysmorphol 2009; 18: 19–23.
Humbert M, Labrune P, Simonneau G : Severe pulmonary arterial hypertension in type 1 glycogen storage disease. Eur J Pediatr 2002; 161: S93–S96.
Wang Y, Oeser JK, Yang C et al: Deletion of the gene encoding the ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein (UGRP)/glucose-6-phosphatase catalytic subunit-beta results in lowered plasma cholesterol and elevated glucagon. J Biol Chem 2006; 281: 39982–39989.
Bandsma RHJ, Prinsen BH, Van Der Velden MDS et al: Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a. Pediatr Res 2008; 63: 702–707.
Rake J, Visser G, Labrune P, Leonard J, Ullrich K, Smit G : Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European study on glycogen storage disease type I (ESGSD I). Eur J Pediatr 2002; 161: S20–S34.
Acknowledgements
The study was funded by the NIHR Manchester Biomedical Research Centre.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Banka, S., Chervinsky, E., Newman, W. et al. Further delineation of the phenotype of severe congenital neutropenia type 4 due to mutations in G6PC3. Eur J Hum Genet 19, 18–22 (2011). https://doi.org/10.1038/ejhg.2010.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.136
Keywords
This article is cited by
-
Severe congenital neutropenia due to G6PC3 deficiency: early and delayed phenotype of a patient
Allergy, Asthma & Clinical Immunology (2023)
-
Hypothesis: A Novel Neuroprotective Role for Glucose-6-phosphatase (G6PC3) in Brain—To Maintain Energy-Dependent Functions Including Cognitive Processes
Neurochemical Research (2020)
-
A Novel Mutation in G6PC3 Gene Associated Non-syndromic Severe Congenital Neutropenia
Indian Pediatrics (2020)
-
Severe congenital neutropenia due to G6PC3 deficiency: early and delayed phenotype in two patients with two novel mutations
Italian Journal of Pediatrics (2014)
-
Testicular failure in a patient with G6PC3 deficiency
Pediatric Research (2014)

{kind=link}