
Abstract
Studies of the fat mass and obesity-associated (FTO) gene provide compelling evidence of genetic variation in the general population that influences fat levels and obesity risk. Studies of the interaction between genetic and environmental factors such as physical activity (PA) will promote the understanding of how lifestyle can modulate genetic contributions to obesity. In this study, we investigated the effect of FTO genotype, and interactions with PA or energy intake, in young children and adolescents. In all, 1–5-year-old children from the Growth, Exercise and Nutrition Epidemiological Study in preSchoolers (GENESIS) study (N=1980) and 11–18-year-old Greek adolescents (N=949) were measured for adiposity-related phenotypes and genotyped at the FTO single-nucleotide polymorphism (SNP) marker, rs17817449. Adolescents were classified as physically active or inactive based on self-reported levels of PA. In adolescents, FTO genotype influenced weight (P=0.001) and BMI (P=0.007). There was also a significant SNP*PA*gender interaction (P=0.028) on BMI, which reflected the association between FTO genotype and BMI in males (P=0.016), but not females (P=0.15), and significant SNP*PA interaction in males (P=0.007), but not females (P=0.74). The FTO genotype effect was more pronounced in inactive than active males. Inactive males homozygous for the G allele had a mean BMI 3 kg/m2 higher than T carriers (P=0.008). In the GENESIS study, no significant association between FTO genotype and adiposity was found. The present findings highlight PA as an important factor modifying the effect of FTO genotype.
Similar content being viewed by others


INTRODUCTION
Childhood obesity is a leading risk factor for the metabolic syndrome and type II diabetes, as well as potentially for subsequent cardiovascular disease in adulthood.1, 2, 3 Obesity has a complex aetiology, but is clearly influenced both by genetic variation and by environmental factors such as diet and lifestyle.4 Physical activity (PA), for example, is recognised as a prevention tool for obesity, is known to increase energy expenditure and also has the potential to regulate appetite,5 although the relationship between PA and obesity is not clearly defined.6
Studies of the recently discovered fat mass and obesity-associated (FTO) gene have provided the most compelling evidence to date of genetic variation in the general population that influences fat levels and risk of obesity.7, 8 A set of single-nucleotide polymorphisms (SNPs) in the first intron of FTO are strongly associated with adiposity/obesity and related phenotypes.7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 It has been shown that adults homozygous for the risk allele at rs9939609 weigh on average ∼3 kg more than those homozygous for the other allele.7 FTO genotype has also been reported to be associated with adiposity in children7, 8, 9 and the strength of its influence to increase during childhood.10
The function of FTO is currently unknown, but is becoming clearer. It is widely expressed throughout the body and highly so in the arcuate nucleus, a region of the hypothalamus involved in regulating energy balance.18 Fischer et al19 reported recently that knocking out Fto in mice results in reduced adiposity through a mechanism apparently involving increased energy expenditure, sympathetic tone and levels of catecholamine hormones, which contrasts with studies to date in human beings that implicate hyperphagia without altered energy expenditure.9, 20, 21, 22, 23
Despite having the largest influence on obesity-related phenotypes of any gene reported thus far, FTO genotype alone only accounts for a small proportion of the variance in these phenotypes. The full influence of FTO will only become clear through more thorough investigation of its interactions with environmental factors such as PA levels. Three studies have reported interactions between PA and FTO genotype in adults, with low activity levels being associated with enhanced genotype effects,24, 25, 26 whereas other studies have reported no interaction with PA.17, 27 However, to the best of our knowledge, no such studies have been conducted in pre-adults. We report here the influence of rs17817449 on adiposity in two cohorts of children, with ages ranging from 1 to 18 years. In adolescent children, we also investigated how PA levels modulate the FTO effects on adiposity-related phenotypes.
METHODS
Subjects
Study participants were those of two previously published cohorts.28, 29 One cohort comprised subjects from the Growth, Exercise and Nutrition Epidemiological Study in preSchoolers (GENESIS),28 which included 1980 healthy children aged 1–5 years from throughout Greece, collected between April 2003 and July 2004. Among them, 52% were male. The other cohort comprised 949 children aged 11–18 years, 53% of whom were male, collected from the Trikala region in central Greece between February 2000 and April 2001.29 Both studies were approved by the ethics committee of the University of Glasgow and by local authorities in Greece. Written informed consent was obtained from the parents of all participants before the study.
Data collection
Data were collected as described below after a period of protocol standardisation in both cohorts. In the GENESIS cohort, all measurements were made as previously described in detail.28 This included basic body composition measurements (weight, height and waist, hip and mid-upper arm circumferences), as well as skinfolds at four sites (biceps, triceps, subscapular and suprailiac). Birth weight data for this cohort were also available from the child health booklet. Energy intake data were collected from all children for 3 days using a combination of weighed food records and 24 h recall or food diaries, as previously described in detail28 and as provided in the Supplementary Material. PA data appropriate for classifying children as active/inactive were not available in the GENESIS cohort. In the adolescent cohort, subjects were measured for height, weight and triceps and subscapular skinfolds as previously described29 and completed a questionnaire documenting their participation in PA both inside and outside of school. Individuals reporting no additional PA outside of the compulsory weekly physical education class at school were classified as inactive (N=289; 98 males and 191 females), whereas those reporting participation in further PA were classified as active (N=660; 401 males and 259 females).29 Inactive individuals displayed higher subscapular skinfolds, tested by GLM adjusted for age and gender, than those classified as active (P=0.02) and displayed a trend towards higher BMI (P=0.075) and triceps skinfold (P=0.077), but not weight (P=0.39). Dietary intake data were not available in the adolescent cohort.
Genotyping and data analysis
SNP variant rs17817449 was genotyped in both cohorts by a PCR-RFLP method, digesting the PCR product with AlwNI restriction enzyme. The forward primer was 5′-CGGTGAAGAGGAGGAGATTG-3′ and the reverse primer 5′-CATCTCTGCCCCAGTTTCTC-3′. The G allele generated an undigested 223 bp product, whereas the T allele yielded 123 and 100 bp fragments after digestion. Genotyping was successful in 94% of subjects in the GENESIS cohort and in 96% of subjects in the adolescent cohort. G allele frequency at rs17817449 was 0.42 in the adolescent cohort and 0.43 in GENESIS. The rs17817449 genotypes were in Hardy–Weinberg equilibrium in both the adolescent (P=0.17) and GENESIS cohorts (P=0.14). rs17817449 is in complete linkage disequilibrium (in European populations) with the more commonly measured SNP rs9939609 (r2=1).
Statistical analysis
Groups were tested for normality using the Ryan–Joiner test, and each phenotype transformed (see Supplementary Material) so as to achieve as close to a normal distribution as possible across each cohort. After transformation, most phenotypes were normal, or approaching normality. Data were analysed using R. We initially investigated the effect of FTO genotype on adiposity phenotypes by ANCOVA. Hypothesising that genotype effects might also differ in age or gender subgroups, we subsequently tested for interactions using a backward stepwise ANCOVA approach. The starting model contained a four-way (SNP*PA*gender*age) interaction term and all hierarchically necessary sub-terms in adolescents and SNP*energy intake (kcal/day)*gender*age in the GENESIS children. Age was classified into single-year age categories. Interaction terms were left in the model if P<0.1. The final model for each cohort consisted of all significant interaction terms and additional main effects not contained within those terms. Post hoc comparisons of phenotypic differences between genotype groups in the adolescent male subgroup were calculated by age-adjusted GLM. To allow for comparison of effects across the age range, age and sex-specific z-scores were calculated for boys and girls separately in single-year age strata in the GENESIS cohort, and in the same manner in adolescents, although 11- and 12-year olds were collapsed into one group, as were 17–18-year olds.
No explicit multiple testing correction was carried out as we view FTO as having high prior likelihood of influencing all phenotypes investigated. In the adolescent cohort, point biserial correlations were calculated between PA status as a dichotomous variable (active or inactive) and adiposity phenotypes. For point biserial correlations, generally r lies between −0.8 and 0.8.30 It was used here to investigate the relationship between PA and adiposity in all subjects, then in risk and protective genotype groups.
RESULTS
Adolescent cohort
In a model-free ANCOVA analysis, FTO genotype was shown to influence weight (P=0.006) and BMI (P=0.02) in the adolescent cohort (Table 1). A genetic model with dominant effects of the T allele (T-dominant model) explained most of the genetic variance, and all subsequent results were derived using this model (ie T carriers are compared with those homozygous for the G allele). Under the T-dominant model, FTO genotype influenced BMI (P=0.007), weight (P=0.001), triceps (P=0.034) and subscapular skinfolds (P=0.021), with GG individuals having higher adiposity than T carriers for all phenotypes (Table 1). As can be seen in Table 1, those homozygous for the G allele had measures of adiposity 0.18–0.27 SD higher than T allele carriers. Interactions between FTO genotype, age, gender and PA on BMI were then investigated. Non-significant terms were removed from the model until the final model included the simplest significant three-way interaction term (SNP*gender*PA: P=0.028) and necessary sub-terms (Supplementary Table 1).
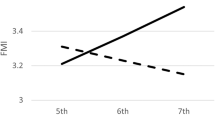
As can be seen in Figure 1, adolescent boys showed differential effects of genotype on BMI in the different PA groups, whereas girls did not. Given the significant SNP*gender*PA interactions, we performed gender-stratified analyses that showed that FTO genotype influenced BMI in males (P=0.016 – in an age-adjusted model not including the SNP*PA interaction term) and that there was a significant SNP*PA interaction in males (P=0.007), but there was neither main effect of FTO genotype on BMI in females (P=0.15), nor any SNP*PA interaction (P=0.74). Subsequent analyses in males, stratified by PA and adjusted for age, showed that inactive males homozygous for the G allele had higher mean BMI than inactive T carriers (P=0.0008), with FTO genotype explaining 10.1% of the variance in BMI among inactive males. These inactive male G homozygotes also had higher weight (P=0.0009) and subscapular skinfolds (P=0.03), while effects on triceps skinfolds tended in the same direction (P=0.07). Inactive male G homozygotes had a mean BMI >3 units (3.25 kg/m2 (95% CI, 1.44–5.05); P=0.001; 0.83 SD) higher than inactive male T carriers. However, when only active males were considered, FTO genotype had no effect on BMI (P=0.52), weight (P=0.32), triceps (P=0.79) or subscapular skinfolds (P=0.85). Age-specific effects of FTO genotype are shown in Supplementary Table 2, in which it can be seen that the direction of effect is consistent throughout the adolescent age range.

Effect of FTO genotype on BMI in adolescents and its interaction with physical activity and gender. Males showed a significant SNP*PA interaction (Pinteraction=0.01 in model-free ANCOVA, as above, and Pinteraction=0.0007 in T-dominant model). Females showed no significant SNP*PA interaction (P=0.54 as above and P=0.74 in T-dominant model). Data are presented as mean ± SEM (adjusted for age). N for each group is as follows: active males – TT: 145, GT: 189, GG: 67; inactive males – TT: 29, GT: 43, GG: 26; active females – TT: 96, GT: 118, GG: 44; inactive females – TT: 61, GT: 91, GG: 39.
Looking at the question the other way round, we asked whether genotype influenced the relationship between PA level and the adiposity phenotypes. Analysis of point biserial correlations between PA status and adiposity phenotypes (Supplementary Table 3) showed weak but significant correlations in the whole cohort, whose magnitude was strengthened in GG homozygotes and weakened in T carriers. For example, the correlation between PA level and BMI was −0.08 in the entire cohort, −0.06 in T carriers, but increased in strength to −0.15 (with P=0.042) in G allele homozygotes.
Genesis cohort (ages 1–5 years)
In the GENESIS cohort, we also tested the association between FTO genotype and energy intake, as well as measures of adiposity: BMI, weight, waist and hip circumference and bicep, tricep, subscapular and suprailiac skinfold, and birth weight. As can be seen in Supplementary Table 4, none of these phenotypes showed any association with FTO genotype after adjustment for age and gender. We further tested for any age, gender or energy intake interactions to establish whether any subgroup effects were being obscured. However, no significant interactions between FTO genotype and energy intake were observed on BMI (P>0.1) using a backward stepwise approach. Furthermore, as can be seen in Supplementary Table 2, only the 3-year-old children showed a significant effect of FTO genotype on age and sex-specific BMI z-scores. Although this was in a direction consistent with the effects in the adolescent cohort, the other age groups showed no significant effects, and directions were inconsistent.
DISCUSSION
We report here that genotype at FTO SNP rs17817449 influences variation in obesity-related measures in adolescents, and that T allele carriers display lower levels of adiposity than G allele homozygotes. We also report an interaction between PA status, gender and FTO genotype, with the obesogenic influence of FTO largely confined to inactive males. In young children, in contrast, there was no association between FTO and adiposity phenotypes or energy intake.
In the adolescent cohort, weight and BMI, adjusted for age, were associated with FTO genotype (P<0.01), whereas skinfold thicknesses showed genotypic differences with P-values slightly higher than this (Table 1). This influence of genotype was strongest when analysed under a T allele-dominant model. In terms of effect size, genotype alone explained 0.4–0.8% of the phenotypic variance for these traits. A significant interaction between FTO genotype and activity status was observed in males and the effect of genotype was stronger when analysed in inactive males, with up to 10% of the variance explained (Table 1c). The proportion of females classed as inactive in this study was greater than that of males (42 vs 20%). It has been shown previously that adolescent females are less active than boys.31 However, if the subjective PA measurement method used in this study is a less specific measure of inactivity in females than males, the inclusion of moderately active females in the inactive group may dilute any interaction, as it appears that the FTO effect is strongest in the least active individuals.17, 26 Nevertheless, the attenuation of the FTO effect by PA is in agreement with previous findings in adults.24, 25, 26 Although some studies have failed to find such an effect,17, 27 a similar trend is in fact visible in the study of Tan et al.17 The equivocal findings may be attributable in part to the subjective measures of PA used in these studies, and further, large studies, using objective monitoring of PA, are required. Inter-population differences in allele frequencies and patterns of linkage disequilibrium may also contribute to heterogeneity of findings. Previous studies have found that the influence of FTO on adiposity phenotypes is apparent from as young as 7 years old7, 32 and that the strength of the association increases into early adolescence.10 In the GENESIS cohort, we observed association neither between FTO genotype and adiposity, which is in line with previous findings reporting no association of FTO genotype and childhood BMI in 2–5-year-old children,33 nor between FTO genotype and energy intake (Supplementary Table 4). However, given the impact of PA on FTO associations in the adolescent cohort, our ability to detect such subgroup-specific genotype effects operating in these younger age groups may be confounded in the absence of useful PA data in the GENESIS cohort.
Obesity phenotypes show a high degree of heritability in familial studies.10 It has also been shown that having one or two overweight parents increases the risk of being overweight in childhood from a very young age,34, 35 further implicating genetic origins for a significant portion of the inter-individual variation in adiposity. However, evidence based on genetic association studies, particularly from genome-wide studies, does not account for a large proportion of this heritability. The FTO gene harbours variation that has the largest effect on adiposity phenotypes of any yet reported, accounting for around 1% of the total phenotypic variance in, for example, BMI in adults.7 Here, we have shown that the influence of FTO may be small overall, but it has a significantly greater influence in the physically inactive portion of the population, with FTO genotype explaining, after age adjustment, as much as 10% of phenotypic variance in inactive adolescent males (Table 1). Studies that do not consider PA levels may well underestimate the potential magnitude of the FTO effect on obesity in the most susceptible groups. Our results support the idea that certain genetic effects may be operating most strongly on groups in a particular environmental milieu and extend the finding that FTO acts most strongly in physically inactive subgroups to the pre-adult life stages. This supports the idea that some genetic influences may be missed in situations where unknown confounding environmental factors are operating to obscure real genetic effects acting on environmentally distinct subgroups.
As well as influencing risk of obesity, our results also suggest that FTO genotype modulates the relationship between adiposity and PA, as the genotype/activity interaction implies that, on one hand, genotypic effects are observed only in certain environments, but also that, on the other, activity effects are different among genotype groups. Increasing PA should have an important function in attaining a negative energy balance by increasing energy expenditure, although the relationship between increasing PA and decreasing obesity is equivocal (reviewed by Wareham et al6). In this study, we find that the weak relationship between PA and obesity is strengthened when examined only in those subjects homozygous for the risk allele (Supplementary Table 3). It must be acknowledged, however, that these findings arise from cross-sectional data with subjective PA measurement and with relatively modest sample size. Although the direction of causality in this relationship is not evident from our cross-sectional data,36 the results do support the use of prospective, longitudinal studies with objective activity measurements to investigate the influence of activity on adiposity further. The influence of activity may have been obscured in previous studies of this type because of the inclusion of genetic ‘non-responders’. Although limited evidence is available from lifestyle intervention studies to date,37, 38 future studies of the relationship between PA and obesity could consider a paradigm involving stratification by FTO genotype to reveal previously obscured environmental effects.
CONCLUSION
It has been suggested that moderate and controlled dietary restriction may prevent FTO-induced obesity9 and previous studies on the genetic influence on obesity have highlighted the importance of genes regulating energy intake.39 However, the finding that PA levels in adolescent males influence the obesogenic effects of FTO risk genotypes highlights both sides of the energy balance equation: intake and expenditure, should be considered in combating obesity. In this study, it was found that FTO genotype had no strong influence in physically active adolescents, but did so in inactive males, with those homozygous for the risk and G allele having a mean BMI as much as 3 kg/m2 higher than those who are physically active or T carriers. This is a promising advert for the benefit of a physically active lifestyle, particularly in those carrying FTO risk variants.
References
Cali AM, Caprio S : Obesity in children and adolescents. J Clin Endocrinol Metab 2008; 93: S31–S36.
Celik T, Iyisoy A, Yuksel UC : Pediatric metabolic syndrome: a growing threat. Int J Cardiol 2008; 142: 302–303.
Weiss R, Dziura J, Burgert TS et al: Obesity and the metabolic syndrome in children and adolescents. N Engl J Med 2004; 350: 2362–2374.
Loos RJ, Bouchard C : FTO: the first gene contributing to common forms of human obesity. Obes Rev 2008; 9: 246–250.
Martins C, Morgan L, Truby H : A review of the effects of exercise on appetite regulation: an obesity perspective. Int J Obes (Lond) 2008; 32: 1337–1347.
Wareham NJ, van Sluijs EM, Ekelund U : Physical activity and obesity prevention: a review of the current evidence. Proc Nutr Soc 2005; 64: 229–247.
Frayling TM, Timpson NJ, Weedon MN et al: A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007; 316: 889–894.
Dina C, Meyre D, Gallina S et al: Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet 2007; 39: 724–726.
Cecil JE, Tavendale R, Watt P et al: An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med 2008; 359: 2558–2566.
Haworth CM, Carnell S, Meaburn EL et al: Increasing heritability of BMI and stronger associations with the FTO gene over childhood. Obesity (Silver Spring) 2008; 16: 2663–2668.
Cornes BK, Lind PA, Medland SE et al: Replication of the association of common rs9939609 variant of FTO with increased BMI in an Australian adult twin population but no evidence for gene by environment (G × E) interaction. Int J Obes (Lond) 2009; 33: 75–79.
Do R, Bailey SD, Desbiens K et al: Genetic variants of FTO influence adiposity, insulin sensitivity, leptin levels, and resting metabolic rate in the Quebec Family Study. Diabetes 2008; 57: 1147–1150.
Hubacek JA, Bohuslavova R, Kuthanova L et al: The FTO gene and obesity in a large eastern European population sample: the HAPIEE study. Obesity (Silver Spring) 2008; 16: 2764–2766.
Hubacek JA, Pitha J, Adamkova V et al: A common variant in the FTO gene is associated with body mass index in males and postmenopausal females but not in premenopausal females. Czech post-MONICA and 3PMFs studies. Clin Chem Lab Med 2009; 47: 387–390.
Scuteri A, Sanna S, Chen WM et al: Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet 2007; 3: e115.
Chang YC, Liu PH, Lee WJ et al: Common variation in the fat mass and obesity-associated (FTO) gene confers risk of obesity and modulates BMI in the Chinese population. Diabetes 2008; 57: 2245–2252.
Tan JT, Dorajoo R, Seielstad M et al: FTO variants are associated with obesity in the Chinese and Malay populations in Singapore. Diabetes 2008; 57: 2851–2857.
Gerken T, Girard CA, Tung YC et al: The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 2007; 318: 1469–1472.
Fischer J, Koch L, Emmerling C et al: Inactivation of the Fto gene protects from obesity. Nature 2009; 458: 894–898.
Speakman JR, Rance KA, Johnstone AM : Polymorphisms of the FTO gene are associated with variation in energy intake, but not energy expenditure. Obesity (Silver Spring) 2008; 16: 1961–1965.
Wardle J, Carnell S, Haworth CM et al: Obesity associated genetic variation in FTO is associated with diminished satiety. J Clin Endocrinol Metab 2008; 93: 3640–3643.
Wardle J, Llewellyn C, Sanderson S et al: The FTO gene and measured food intake in children. Int J Obes (Lond) 2009; 33: 42–45.
Timpson NJ, Emmett PM, Frayling TM et al: The fat mass- and obesity-associated locus and dietary intake in children. Am J Clin Nutr 2008; 88: 971–978.
Rampersaud E, Mitchell BD, Pollin TI et al: Physical activity and the association of common FTO gene variants with body mass index and obesity. Arch Intern Med 2008; 168: 1791–1797.
Andreasen CH, Stender-Petersen KL, Mogensen MS et al: Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes 2008; 57: 95–101.
Vimaleswaran KS, Li S, Zhao JH et al: Physical activity attenuates the body mass index-increasing influence of genetic variation in the FTO gene. Am J Clin Nutr 2009; 90: 425–428.
Jonsson A, Renstrom F, Lyssenko V et al: Assessing the effect of interaction between an FTO variant (rs9939609) and physical activity on obesity in 15 925 Swedish and 2511 Finnish adults. Diabetologia 2009; 52: 1334–1338.
Manios Y : Design and descriptive results of the ‘Growth, Exercise and Nutrition Epidemiological Study in preSchoolers’: the GENESIS study. BMC Public Health 2006; 6: 32.
Moran CN, Vassilopoulos C, Tsiokanos A et al: Effects of interaction between angiotensin I-converting enzyme polymorphisms and lifestyle on adiposity in adolescent Greeks. Obes Res 2005; 13: 1499–1504.
Karabinus RA : The r-point biserial limitation. Educ Psychol Meas 1975; 35: 277–282.
Brodersen NH, Steptoe A, Boniface DR et al: Trends in physical activity and sedentary behaviour in adolescence: ethnic and socioeconomic differences. Br J Sports Med 2007; 41: 140–144.
Hakanen M, Raitakari OT, Lehtimaki T et al: FTO genotype is associated with body mass index after the age of seven years but not with energy intake or leisure-time physical activity. J Clin Endocrinol Metab 2009; 94: 1281–1287.
Zhao J, Bradfield JP, Li M et al: The role of obesity-associated loci identified in genome-wide association studies in the determination of pediatric BMI. Obesity (Silver Spring) 2009; 17: 2254–2257.
Manios Y, Costarelli V, Kolotourou M et al: Prevalence of obesity in preschool Greek children, in relation to parental characteristics and region of residence. BMC Public Health 2007; 7: 178.
Magarey AM, Daniels LA, Boulton TJ et al: Predicting obesity in early adulthood from childhood and parental obesity. Int J Obes Relat Metab Disord 2003; 27: 505–513.
Ekelund U, Brage S, Besson H et al: Time spent being sedentary and weight gain in healthy adults: reverse or bidirectional causality? Am J Clin Nutr 2008; 88: 612–617.
Rankinen T, Rice T, Teran-Garcia M et al: FTO genotype is associated with exercise training-induced changes in body composition. Obesity (Silver Spring) 2009; 18: 322–326.
Lappalainen TJ, Tolppanen AM, Kolehmainen M et al: The common variant in the FTO gene did not modify the effect of lifestyle changes on body weight: the Finnish Diabetes Prevention Study. Obesity (Silver Spring) 2009; 17: 832–836.
Willer CJ, Speliotes EK, Loos RJ et al: Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 2009; 41: 25–34.
Acknowledgements
The GENESIS study was supported by a research grant from Friesland Foods Hellas. The adolescent Greek study was funded in part by a BBSRC research grant no. 17/D17566. The co-operation of all schools, subjects and their families is greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
YM works as a part-time scientific consultant for Friesland Foods Hellas. This sponsor had no role or voice in the study design, data collection, analysis or writing of the article. The other authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Scott, R., Bailey, M., Moran, C. et al. FTO genotype and adiposity in children: physical activity levels influence the effect of the risk genotype in adolescent males. Eur J Hum Genet 18, 1339–1343 (2010). https://doi.org/10.1038/ejhg.2010.131
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.131
Keywords
This article is cited by
-
Multiple novel gene-by-environment interactions modify the effect of FTO variants on body mass index
Nature Communications (2016)
-
The Epidemiology of Obesity: A Big Picture
PharmacoEconomics (2015)
-
Behavioral and Environmental Modification of the Genetic Influence on Body Mass Index: A Twin Study
Behavior Genetics (2015)
-
The ‘Fat Mass and Obesity Related’ (FTO) gene: Mechanisms of Impact on Obesity and Energy Balance
Current Obesity Reports (2015)
-
Modification of genetic influences on adiposity between 36 and 63 years of age by physical activity and smoking in the 1946 British Birth Cohort Study
Nutrition & Diabetes (2014)
