
Abstract
Oculofaciocardiodental (OFCD) and Lenz microphthalmia syndromes form part of a spectrum of X-linked microphthalmia disorders characterized by ocular, dental, cardiac and skeletal anomalies and mental retardation. The two syndromes are allelic, caused by mutations in the BCL-6 corepressor gene (BCOR). To extend the series of phenotypes associated with pathogenic mutations in BCOR, we sequenced the BCOR gene in patients with (1) OFCD syndrome, (2) putative X-linked (‘Lenz’) microphthalmia syndrome, (3) isolated ocular defects and (4) laterality phenotypes. We present a new cohort of females with OFCD syndrome and null mutations in BCOR, supporting the hypothesis that BCOR is the sole molecular cause of this syndrome. We identify for the first time mosaic BCOR mutations in two females with OFCD syndrome and one apparently asymptomatic female. We present a female diagnosed with isolated ocular defects and identify minor features of OFCD syndrome, suggesting that OFCD syndrome may be mild and underdiagnosed. We have sequenced a cohort of males diagnosed with putative X-linked microphthalmia and found a mutation, p.P85L, in a single case, suggesting that BCOR mutations are not a major cause of X-linked microphthalmia in males. The absence of BCOR mutations in a panel of patients with non-specific laterality defects suggests that mutations in BCOR are not a major cause of isolated heart and laterality defects. Phenotypic analysis of OFCD and Lenz microphthalmia syndromes shows that in addition to the standard diagnostic criteria of congenital cataract, microphthalmia and radiculomegaly, patients should be examined for skeletal defects, particularly radioulnar synostosis, and cardiac/laterality defects.
Similar content being viewed by others


Introduction
The X-linked microphthalmia syndromes, 11 of which are currently defined (Table 1), comprise a clinically and molecularly diverse group of disorders, a number of which overlap with X-linked mental retardation syndromes. Lenz microphthalmia syndrome has been recognized for more than 50 years and was previously assumed to be a unitary entity. However, clinical and linkage analyses suggest that it may be aetiologically heterogeneous, linked to both Xq27-q281 (MCOPS1; MIM 309800) or Xp11.42, 3 (MCOPS2; MIM 300166). Specifically, the MCOPS2 form of Lenz microphthalmia syndrome has been shown to be caused by mutation of the BCL-6 corepressor gene (BCOR);3 the genetic aetiology of MCOPS1 remains unknown. The original report of Lenz described a family with variable ocular manifestations, including anophthalmia, microphthalmia and coloboma. There were numerous extraocular anomalies including mental retardation, palatal and dental anomalies, congenital heart defects, skeletal defects (affecting the fingers and clavicles), unilateral renal aplasia and cryptorchidism;4 this phenotype substantially overlaps with other X-linked microphthalmia syndromes (Table 1). Another X-linked microphthalmia-associated condition, oculofaciocardiodental (OFCD) syndrome, is characterized by ocular defects (congenital cataracts, microphthalmia), facial anomalies (septate nasal tip, high nasal bridge, midface hypoplasia, palatal anomalies), congenital cardiac defects (atrial/ventricular septal defects, other complex heart defects), dental irregularities (canine radiculomegaly, delayed and persistent dentition, hypodontia) and skeletal anomalies (syndactyly, hammer-type flexion deformities).5, 6, 7, 8, 9, 10 All affected individuals are female, with several incidences of mother-daughter transmission.
In 2004, a sequence variant was identified within the BCOR gene in a single family with Lenz microphthalmia syndrome.3 The missense mutation (c.254C>T, p.P85L) cosegregated with the disease phenotype and was not identified in more than 450 control chromosomes. It represents the sole molecular cause of Lenz microphthalmia syndrome identified to date. In the same study, BCOR mutations were found in all tested patients with OFCD syndrome, suggesting the two conditions were allelic. The mutation types included nonsense, frameshift, deletion and splicing mutations, a finding subsequently replicated by others. Importantly, OFCD syndrome has been shown to encompass defects of laterality, including the heart and other viscera, suggesting that BCOR is necessary for left–right asymmetric development and that mutations in BCOR may represent a source of the reported male excess of laterality defects.11, 12
This study aims to expand our understanding of the phenotypes associated with mutations in BCOR, supporting the hypothesis that OFCD syndrome is solely associated with null mutations in BCOR and to identify additional patients with Lenz microphthalmia syndrome who carry mutations in BCOR. We have demonstrated that mutations in BCOR may be responsible for apparently isolated ocular anomalies and tested the hypothesis that BCOR mutations cause non-syndromic cardiac/laterality defects. The continuous accumulation of patient data allows the frequency of non-cardinal phenotypes to be estimated and improve diagnosis and treatment of patients.
Materials and methods
Direct sequencing of the BCOR gene
The coding exons and flanking intronic sequences of the BCOR gene were amplified by PCR and directly sequenced as previously reported.3
Semi-quantitative multiplex fluorescent PCR analysis of the BCOR gene
Deletions within the BCOR gene were detected by semi-quantitative multiplex fluorescent PCR (QMF-PCR).13, 14 All exons of the BCOR gene and three control genes (DSCRI – chromosome 21, CFTR – chromosome 7 and Factor IX – chromosome X) were amplified in two duplicate multiplex reactions (primer sequences in Supplementary Table 1). One of each primer pair was labelled with the fluorescent phosphoramidite 6-FAM dye. Amplifications were performed in 25 μl reactions using the QIAGEN Multiplex PCR kit (Qiagen, France) with 75 ng of genomic DNA and a mix of primers (concentration range 1.3–8 μ M). The reaction started with an initial denaturation of 15 min at 95°C followed by 23 cycles at 95°C for 30 s/60°C for 30 s/72°C for 45 s with an increment of 3 s per cycle. Final extension was at 72°C for 10 min. PCR products were purified and processed as previously described.14 Two control DNAs (male and female) were included in each experiment. Results were analysed by superimposing fluorescent profiles of tested patients and controls.
Fluorescent in situ hybridization analysis of the BCOR gene
To determine the proportion of cells with a deletion in the BCOR gene, fluorescent in situ hybridization (FISH) was performed as previously described.15 Metaphase spreads of peripheral leucocytes were obtained according to standard techniques. BAC clones used in FISH experiments were provided by The Wellcome Trust Sanger Institute (Cambridge, UK). Clones localized on chromosome Xp11.4 (RP11-320G24, RP11-330L22 and RP11-429N5) were directly labelled with Cy3. Chromosomes were counterstained with DAPI. The specific signal intensity and its sublocalization along the chromosome axis were analysed using a Leica fluorescence microscope equipped with the Visilog-6 program (Noesis, Les Ulis, France).
Ethics approval
The human subject research described here was reviewed and approved by ethics committees at St Mary's Hospital (Manchester, UK), the National Institutes of Health (Bethesda, MD, USA), Baylor College of Medicine (Houston, TX, USA), and Hôpital Henri Mondor (Créteil, France).
Results
Sequence and copy number analysis of BCOR in patients with OFCD syndrome
From existing literature and previous work, we have identified 33 females with OFCD syndrome.3, 5, 6, 7, 8, 9, 10, 11, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 In this study, two of these patients are described in further clinical detail11 and mutational analysis of BCOR completed in a family (mother and daughter).25 In addition to these four previously reported patients, we have identified a further 31 females putatively diagnosed with OFCD syndrome and found mutations in BCOR in all families, bringing the cohort of patients described here to 35 cases. A summary of the phenotypes and mutations identified in patients with OFCD syndrome is given in Table 2. The positions of mutations in relation to the exon sequences are shown in Figure 1. The reference sequence used for mutation numbering is given in Supplementary Data 1. The 31 novel patients described here brings the total number of reported cases of OFCD syndrome to 64. We have also sequenced three females with features of OFCD syndrome who proved negative for BCOR mutations; however, the phenotypes were atypical for OFCD syndrome, with a notable lack of radiculomegaly in the dental phenotype in all cases.

Location of reported mutations within the BCOR gene. Arrowheads above the diagram indicate mutations described in previous reports. Mutations indicated below the diagram are those identified in this study (see Table 2). Arrowheads in white indicate mutations corresponding to females with OFCD syndrome. Arrowheads in grey indicate mutations corresponding to males with Lenz microphthalmia syndrome. Arrowhead in black indicates an in-frame deletion occurring in a female with OFCD syndrome. Deletions are indicated by solid horizontal lines.
The mutations identified here and in other studies involve deletions of significant portions of the BCOR coding sequence, alterations to conserved splice acceptor sites (predicted to lead to exon skipping with concomitant frameshifts) or small insertions/deletions which cause frameshifts and generate premature stop codons; it is predicted that such premature stop codons will trigger nonsense-mediated decay of the mRNA although the generation of C-terminally truncated protein species is also possible. The observation that, in all females tested to date, X-inactivation is grossly skewed in favour of the wild-type allele (data not shown) suggests that loss of wild-type BCOR protein confers significant selective disadvantage, certainly in haematological lineages. Females with OFCD syndrome are therefore functional mosaics, with cell populations and tissues expressing either wild-type BCOR (where lack of BCOR function is presumed lethal) or no BCOR/truncated BCOR (where lack of BCOR function can be supported). Amongst the pathogenic variants are two further instances of a previously reported c.2926C>T p.R976X nonsense mutation.3
In this study, we report for the first time two families with individuals mosaic for a mutation in BCOR (OFCD XVI and OFCD XVII). In OFCD XVI, the individuals mosaic for a BCOR mutation have the cardinal phenotype associated with OFCD syndrome. In contrast, the mosaic individual from OFCD XVII is asymptomatic. In both families, offspring present with the cardinal OFCD syndrome phenotype and are non-mosaic for heterozygous BCOR mutations.
OFCD XVI
Individuals XVI.1 and XVI.2 are monozygotic twin sisters, both presenting with a classical OFCD syndrome phenotype. Individual XVI.3 (the daughter of individual XVI.1) also presented with an overlapping array of features. Using a quantitative fluorescent PCR method, a deletion of at least exons 4–15 was detected in patients XVI.1 and XVI.2. However, the decrease in PCR product amount derived from the BCOR gene was estimated at around 75% of control peak height, rather than the 50% expected for a non-mosaic deletion (Figure 2a and b). This result was recapitulated for all BCOR fragments amplified and strongly suggested somatic mosaicism of this deletion. By contrast, amplification was reduced to 50% of control peak height in patient XVI.3, suggesting non-mosaic distribution of the BCOR deletion (Figure 2c). FISH analysis was undertaken to confirm somatic mosaicism in individuals XVI.1 and XVI.2. FISH analysis on metaphase chromosomes from both patients demonstrated a deletion corresponding to BAC clone RP11-330L22 in 52/100 metaphase preparations for each patient, with only one signal from the X chromosome (Figure 2g; data from patient XVI.1 not shown). Two distinct signals were detected in the remaining 48 leucocyte preparations (Figure 2h; data from patient XVI.1 not shown). X chromosomes were identified using an X centromere marker and by banding pattern (not shown). These results supported the hypothesis that these two individuals are mosaic for the deleted region. For patient XVI.3, 100/100 metaphase preparations generated a single signal, confirming the non-mosaic nature of the deletion in this patient (Figure 2i). Two BAC clones flanking RP11-330L22, RP11-429N5 (proximal) encompassing exons 4–15 of BCOR and RP11-320G24 (distal) were tested in patient XVI.3. Two specific signals were detected in each metaphase preparation, suggesting that these regions are not deleted in this family (the proximal RP11-429N5 probe contains sufficient sequence outside of the BCOR gene to permit hybridization).

Evidence for somatic mosaicism in individuals XVI.1, XVI.2 and XVII.1. (a–e) QMF-PCR fluorescent spectra obtained for fragments of the BCOR gene and control genes, with patient peaks in red and control subject peaks in blue. Patient XVI.1 (a), patient XVI.2 (b) and patient XVII.1 (d) display diminished peak intensity to approximately 75% of the control peak intensity, suggesting mosaicism of the deleted region. Patient XVI.3 (c) and patient XVII.2 (e) display diminished peak intensity to approximately 50% of the control peak intensity, suggesting a non-mosaic deletion. (f) Sequence chromatogram of patient XVII.2, confirming a deletion between introns 12 and 14, resulting in loss of exons 13 and 14. (g–i) FISH analysis using BAC clone RP11-330L22, encompassing the BCOR gene, with signals indicated by white arrows. Data for patient XVI.1 are equivalent to patient XVI.2. Patient XVI.2 (g, h) displays a single signal in approximately 50% of metaphase preparations (g) whereas the remaining preparations show two specific signals (h), confirming that these individuals are mosaic for a deletion of the BCOR gene. For patient XVI.3 (i), a specific single signal was observed in 100% of metaphase preparations, confirming that the deletion of the BCOR gene is non-mosaic and present in all cells.
OFCD XVII
The proband (XVII.2), a 14–month-old girl, presented with a classical OFCD syndrome phenotype. The mother was asymptomatic and had a normal panoramic dental X-ray. Quantitative fluorescent PCR detected a reduced amount of product corresponding to exons 13 and 14, suggesting a deletion of these exons in both females. Comparison of amounts of PCR product obtained suggested that the mother (XVII.1) was mosaic for this deletion, with the amount of PCR product at 75% of the control peak height (Figure 2d). Amplification from exons 8 and 9 were at the control intensity. Amplification from proband XVII.2 was reduced to 50% for exons 13 and 14, suggesting a non-mosaic distribution of this deletion (Figure 2e). PCR amplification and sequencing of a region spanning the proposed deletion region was performed to refine the deletion to 1410 bp from intron 12–14 (Figure 2f).
Clinical features of OFCD syndrome
The cohort presented here comprises 31 new females and four previously described females with mutations in BCOR (35 patients). However, one female (XVII.1) is asymptomatic, despite proving mosaic for a BCOR mutation, and thus is excluded from the phenotypic evaluation, bringing the cohort of symptomatic females to 34. A summary of the frequency of phenotypes within the 34 symptomatic females is given in Table 3.
Ocular
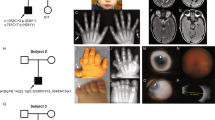
OFCD syndrome is associated with ocular defects and the cardinal manifestation of congenital cataract is present in each individual in the new cohort presented here (34/34). In addition to congenital cataract, patients displayed an array of eye anomalies, most frequently microphthalmia and/or microcornea in 82% of patients (28/34; Figure 3a and b).

OFCD syndrome. (a, b) Typical facial features of OFCD syndrome include microphthalmia (left eye of each patient), long narrow facies and septate nasal cartilage. (c) The dental phenotype of OFCD syndrome includes hypodontia and tooth fusion and duplication. (d, f) Digit anomalies in OFCD syndrome include second-third toe syndactyly (d) and hammer toes (e). (f) Radioulnar synostosis in a patient with OFCD syndrome.
Facial
In our cohort, 31 patients were examined for facial anomalies and a facial phenotype was recorded in 26 patients. The most specific facial manifestation is septate nasal cartilage, observed in 96% (25/26) of affected patients (Figure 3a and b), often associated with a high nasal bridge and long and/or narrow face. Many of the patients had palatal anomalies, including cleft palate, high-arched palate and bifid uvula (31%; 8/26).
Cardiac
Where examined, the majority of patients in the cohort presented here have a congenital heart defect (74%; 20/27), with 85% (17/20) presenting with a septal defect, most commonly an atrial septal defect. Further cardiac anomalies such as pentalogy of Fallot, double-outlet-right-ventricle, valve insufficiencies and patent ductus arteriosus are observed, suggesting a role for BCOR in multiple cardiac processes.
Dental
In this cohort, 30 patients were examined for dental defects. Owing to age, data corresponding only to primary dentition were available for eight patients. Of these, 75% (6/8) have a dental phenotype of delayed primary dentition and/or persistent primary teeth. Two patients have thus far unaffected primary dentition but it is anticipated that secondary dentition will be affected. Data regarding both primary and secondary dentition are available for 22 patients in this cohort and all have a variety of dental anomalies associated with OFCD syndrome, that is, delayed/persistent dentition with multiple unerupted teeth, root radiculomegaly and absent/duplicated/fused teeth (100%; 22/22; Figure 3c). Root radiculomegaly, a unique and cardinal diagnostic feature of OFCD syndrome, is present in 91% (20/22) of patients, with diagnosis limited in the remaining two patients by the unavailability of a dental X-ray.
Skeletal
A number of abnormalities of the skeleton have been reported to be associated with OFCD syndrome and in 29 patients examined, we observe skeletal anomalies in 28 cases. Hammer toes are present in 54% (15/28) of affected patients (Figure 3e) and second-third toe syndactyly in 57% (16/28; Figure 3d); 93% (26/28) of patients have at least one of these phenotypes. Radioulnar synostosis is more recently recognized as a skeletal feature of OFCD syndrome and was detected in 25% (7/28) of affected patients of the cohort described here (Figure 3f), with a further two patients reporting limited supination.
Other
Developmental problems have been observed in patients with OFCD syndrome and in the cohort presented here, 18% (6/34) of patients demonstrate a level of mental retardation, although in most cases this is mild. Hearing loss, both conductive and sensorineural, is observed in 15% (5/34) of patients. Patients with OFCD syndrome may manifest gastrointestinal problems, possibly as part of a laterality phenotype, and we report feeding difficulties and repeated vomiting and reflux in 9% (3/34) of the cohort presented here. A further manifestation of defective lateral patterning, asplenia, is observed in one patient.
Sequence analysis of BCOR in males with presumed X-linked (‘Lenz’) microphthalmia syndrome
To date, a single family in which males are affected by X-linked microphthalmia remain the sole patients with ‘Lenz’ microphthalmia syndrome to have been shown to carry a BCOR mutation.3 We have sequenced the BCOR gene in 21 males with a putative diagnosis of Lenz microphthalmia syndrome, based on an ocular phenotype presenting with additional features of Lenz microphthalmia syndrome, including mental retardation, hearing impairment and skeletal/digital anomalies. In the majority of cases (20/21), there was no evidence of a family history.
We identified a missense mutation in one patient with Lenz microphthalmia syndrome, c.254C>T, predicting p.P85L substitution at the protein level (Lenz I; Table 2). This missense change is identical to the mutation previously described to be associated with Lenz microphthalmia syndrome.3 The patient presented with bilateral microphthalmia, mild mental retardation, atrial septal defect and a typical array of skeletal and digital abnormalities. Interestingly, the patient presented with right radioulnar synostosis, a phenotype not previously associated with Lenz microphthalmia syndrome but strongly linked to OFCD syndrome.
We did not find a mutation in BCOR in 20 of the male patients, suggesting that (1) the diagnosis of Lenz microphthalmia syndrome is disputed or (2) mutations in BCOR are not the major cause of Lenz microphthalmia syndrome, a finding replicated by others.22
Sequence analysis of BCOR in patients with unspecified ocular developmental anomalies
To investigate the contribution of mutations in BCOR in non-specific forms of ocular and mental retardation syndromes, we analysed the BCOR gene in a panel of 96 patients with isolated microphthalmia, coloboma and/or mental retardation. We identified a single mutation in BCOR in a female patient with what had been recorded as isolated bilateral cataract and unilateral microphthalmia (OFCD VII.1). The proband's mother had a similar phenotype. The mutation was c.4303_4307delCCATG p.P1435LfsX24. On further questioning, it was found that the patient had numerous primary teeth removed in teenage years and had second-third toe syndactyly, suggesting that this female has a mild OFCD syndrome phenotype.
Sequence analysis of BCOR in patients with unspecified lateral specification defects
We have previously identified defects in lateral specification as part of the clinical spectrum of OFCD syndrome.11 We therefore hypothesize that mutations in BCOR may account for non-syndromic laterality defects, with heart defects representing the most common manifestation of defects in lateral determination. This hypothesis is supported by the observation that in population studies, there is a ∼2:1 male predominance of patients with laterality defects not attributable to mutations in ZIC3;12, 26 further X-linked genes, such as BCOR, may therefore be involved in human lateral specification. We analysed the BCOR gene in a panel of 96 patients with a variety of cardiac/laterality defects. We identified sequence alterations in three patients: a female with situs inversus and dextrocardia (heterozygous, c.2288G>T, p.R763L), a female with mesocardia, VSD, partial anomalous pulmonary venous return and intestinal malrotation (heterozygous, c.3974A>G, p.K1325R) and a male with transposition of great arteries, pulmonary atresia, VSD and asplenia (hemizygous, c.3974A>G, p.K1325R). These sequence alterations do not appear in SNP databases or within the available EST sequences. However, these changes were also identified in three sequences from a panel of 171 ethnically matched unaffected controls (one female heterozygous for c.2288G>T, p.R763L; two females heterozygous for c.3974A>G, p.K1325R), suggesting that they are rare polymorphisms rather than pathogenic changes.
Discussion
In this study, we have focused on the precise clinical features of OFCD and Lenz microphthalmia syndromes, allelic disorders caused by mutation of the BCOR gene, as examples of X-linked syndromic microphthalmia-associated conditions. Given the heterogeneity amongst X-linked microphthalmia syndromes, extension of our knowledge of the mechanism by which BCOR mutation is associated with OFCD and Lenz microphthalmia syndromes may be pertinent to other disorders. To that end, we searched for mutations in the BCOR gene in patients with OFCD syndrome, presumed X-linked recessive (‘Lenz’) microphthalmia syndrome, isolated ocular defects and lateral specification defects.
Amongst pathogenic sequence variants that cause OFCD syndrome and Lenz microphthalmia syndromes, the mutations presented here confirm the sharply demarcated genotype/phenotype correlation whereby hemizygous males that carry a missense mutation in BCOR have Lenz microphthalmia syndrome and heterozygous females carrying a null allele have OFCD syndrome. The phenotype in female patients may be variable, not only due to differential X-inactivation effects, but also due to somatic mosaicism, a novel finding in three females in the cohort presented here. To date, mutations in BCOR are the sole molecular cause of OFCD syndrome and all mutations identified are null alleles. It is notable that females with putative diagnoses of OFCD syndrome in the absence of radiculomegaly prove to be negative for mutations in BCOR, suggesting a strong association of this cardinal phenotype with mutations in BCOR. The mutation we found in a female patient with apparently isolated microphthalmia and cataract suggests that OFCD syndrome may be under diagnosed and, at the mild end of the spectrum, may have relatively insignificant non-ocular features.
Three patients with OFCD syndrome with manifestations consistent with defective lateral patterning (dextrocardia, asplenia and intestinal malrotation) have been described.11 The clinical features of two of these patients (II.1 and III.6), who display dextrocardia and asplenia respectively, are described in more detail in this report. As it has been demonstrated that BCOR is necessary in the frog embryo to confer correct lateral organization of heart and intestinal tract, it is hypothesized that defects of lateral patterning, particularly of internal organs, are a feature of OFCD syndrome. The association of cardiac septal defects and correct laterality specification is well documented and it is likely that the high frequency of cardiac septal defects in OFCD syndrome is the most common manifestation of aberrant laterality specification. Defects of the midline are aetiologically linked with lateral patterning and it is possible that the facial clefting and septate nasal cartilage observed in OFCD syndrome also result from defective lateral patterning.
Amongst the skeletal features associated with OFCD syndrome it is of note that radioulnar synostosis, a more recently recognized feature, was detected in 25% of the patients with a skeletal phenotype described here. This was also a feature of the patient with Lenz microphthalmia syndrome with the c.254C>T, p.P85L missense mutation. This is identical to what was previously described in Lenz microphthalmia syndrome,3 supporting the pathogenicity of this sequence variant and also suggesting that radioulnar synostosis is an important marker of BCOR mutation.
The genetic causes of Lenz microphthalmia syndrome thus remain largely undefined. Subsequent to the first report by Lenz (1955),4 there have been a large number of reports of X-linked recessive microphthalmia syndrome.27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41 These reports show broad phenotypic variability in males with multiple congenital anomalies. However, in some cases, the absence of a proven X-linked family history must call into question the validity of the diagnosis. It is likely that some reported cases of ‘sporadic Lenz microphthalmia syndrome’ are in fact misdiagnosed and represent other forms of ‘syndromic microphthalmia’. As a result, the exact incidence of Lenz microphthalmia syndrome remains uncertain although the number of families with proven X-linked inheritance is very small. Unfortunately, a genotypic diagnosis of Lenz microphthalmia syndrome is currently impossible due to lack of genetic information. Lenz microphthalmia syndrome has been associated with at least two genetic loci, one at Xq27-q281 and the second occupied by the BCOR gene.2, 3 After exclusion of a BCOR mutation in all except one of our male cohort, we examined other candidate genes which might be associated with X-linked microphthalmia/mental retardation, specifically PQBP1 (mutations in which cause microphthalmia and mental retardation)42 and BCOR-like 1 (based on homology to BCOR and chromosomal location at Xq26.1, in the region of the MCOPS1 locus). No mutations were found in either gene (data not shown), demonstrating that neither is a major cause of X-linked microphthalmia syndromes. Combined with the low incidence of mutations in BCOR in male microphthalmia patients, these data suggest that the major locus for X-linked microphthalmia/mental retardation remains unidentified.
In summary, we have undertaken careful phenotypic analysis of both females with OFCD syndrome and male patients with MCOPS2. Apart from ocular defects, facial dysmorphia, congenital heart defects and dental anomalies, we find a high incidence of skeletal defects. Although both congenital heart defects and defects of laterality determination may be associated with OFCD, we did not find BCOR sequence variants amongst those with unspecified lateral specification defects. Nonetheless we recommend that laterality defects should be investigated, both as cardiac manifestations and intestinal problems in patients with OFCD syndrome. Finally, we suggest that the skeletal phenotype of radioulnar synostosis – which we have seen to be associated with both OFCD and Lenz microphthalmia syndrome – as a further clinical predictor of BCOR mutation.
References
Forrester S, Kovach MJ, Reynolds NM, Urban R, Kimonis V : Manifestations in four males with and an obligate carrier of the Lenz microphthalmia syndrome. Am J Med Genet 2001; 98: 92–100.
Ng D, Hadley DW, Tifft CJ, Biesecker LG : Genetic heterogeneity of syndromic X-linked recessive microphthalmia-anophthalmia: is Lenz microphthalmia a single disorder? Am J Med Genet 2002; 110: 308–314.
Ng D, Thakker N, Corcoran CM et al: Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet 2004; 36: 411–416.
Lenz W : Recessive, sex-limited microphthalmia with multiple abnormalities. Z Kinderheilkd 1955; 77: 384–390.
Hayward JR : Cuspid gigantism. Oral Surg Oral Med Oral Pathol 1980; 49: 500–501.
Marashi AH, Gorlin RJ : Radiculomegaly of canines and congenital cataracts – a syndrome? Oral Surg Oral Med Oral Pathol 1990; 70: 802–803.
Marashi AH, Gorlin RJ : Radiculomegaly of canine teeth and congenital cataracts: confirmation of a syndrome. Am J Med Genet 1992; 42: 143.
Wilkie AO, Taylor D, Scambler PJ, Baraitser M : Congenital cataract, microphthalmia and septal heart defect in two generations: a new syndrome? Clin Dysmorphol 1993; 2: 114–119.
Aalfs CM, Oosterwijk JC, van Schooneveld MJ, Begeman CJ, Wabeke KB, Hennekam RC : Cataracts, radiculomegaly, septal heart defects and hearing loss in two unrelated adult females with normal intelligence and similar facial appearance: confirmation of a syndrome? Clin Dysmorphol 1996; 5: 93–103.
Gorlin RJ, Marashi AH, Obwegeser HL : Oculo-facio-cardio-dental (OFCD) syndrome. Am J Med Genet 1996; 63: 290–292.
Hilton EN, Manson FD, Urquhart JE et al: Left-sided embryonic expression of the BCL-6 corepressor, BCOR, is required for vertebrate laterality determination. Hum Mol Genet 2007; 16: 1773–1782.
Lin AE, Ticho BS, Houde K, Westgate MN, Holmes LB : Heterotaxy: Associated conditions and hospital-based prevalence in newborns. Genet Med 2000; 2: 157–172.
Niel F, Martin J, Dastot-Le-Moal F et al: Rapid detection of CFTR gene rearrangements impacts on genetic counselling in cystic fibrosis. J Med Genet 2004; 41: e118.
Yau SC, Bobrow M, Mathew CG, Abbs SJ : Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J Med Genet 1996; 33: 550–558.
Pinkel D, Straume T, Gray JW : Cytogenetic analysis using quantitative, high-sensitivity fluorescence hybridisation. Proc Natl Acad Sci USA 1986; 83: 2934–2938.
Opitz C, Horn D, Lehmann R, Dimitrova T, Fasmers-Henke K : Oculo-facio-cardio-dental (OFCD) syndrome. J Orofac Orthop 1998; 59: 178–185.
Schulze BR, Horn D, Kobelt A, Tariverdian G, Stellzig A : Rare dental abnormalities seen in oculo-facio-cardio-dental (OFCD) syndrome: three new cases and review of nine patients. Am J Med Genet 1999; 82: 429–435.
Barthelemy I, Samuels L, Kahn DM, Schendel SA : Oculo-facio-cardio-dental syndrome: two new cases. J Oral Maxillofac Surg 2001; 59: 921–925.
Kawamoto T, Motohashi N, Ohyama K : A case of oculo-facio-cardio-dental syndrome with integrated orthodontic-prosthodontic treatment. Cleft Palate Craniofac J 2004; 41: 84–94.
Hedera P, Gorski JL : Oculo-facio-cardio-dental syndrome: skewed X chromosome inactivation in mother and daughter suggest X-linked dominant Inheritance. Am J Med Genet 2003; 123: 261–266.
Tsukawaki H, Tsuji M, Kawamoto T, Ohyama K : Three cases of oculo-facio-cardio-dental (OFCD) syndrome. Cleft Palate Craniofac J 2005; 42: 467–476.
Horn D, Chyrek M, Kleier S et al: Novel mutations in BCOR in three patients with oculo-facio-cardio-dental syndrome, but none in Lenz microphthalmia syndrome. Eur J Hum Genet 2005; 13: 563–569.
Oberoi S, Winder AE, Johnston J, Vargervik K, Slavotinek AM : Case reports of oculofaciocardiodental syndrome with unusual dental findings. Am J Med Genet 2005; 136: 275–277.
Turkkahraman H, Sarioglu M : Oculo-facio-cardio-dental syndrome: report of a rare case. Angle Orthod 2006; 76: 184–186.
McGovern E, Al-Mudaffer M, McMahon C, Brosnahan D, Fleming P, Reardon W : Oculo-facio-cardio-dental syndrome in a mother and daughter. Int J Oral Maxillofac Surg 2006; 35: 1060–1062.
Ware SM, Peng JL, Zhu LR et al: Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet 2004; 74: 93–105.
Hoefnagel D, Keenan ME, Allen FH : Heredofamilial bilateral anophthalmia. Arch Ophthalmol 1963; 69: 760–764.
Goldberg MF, McKusick VA : X-linked colobomatous microphthalmos and other congenital anomalies: a disorder resembling Lenz's dysmorphogenetic syndrome. Am J Ophthalmol 1971; 71: 1128–1133.
Ogunye OO, Murray RF, Osgood T : Linkage studies in Lenz microphthalmia. Hum Hered 1975; 25: 493–500.
Baraitser M, Winter RM, Taylor DS : Lenz microphthalmia – a case report. Clin Genet 1982; 22: 99–101.
Glanz A, Forse A, Polomenom RC, Cole DE : Lenz microphthalmia: a malformation syndrome with variable expression of multiple congenital anomalies. Can J Ophthalmol 1983; 18: 41–44.
Pallotta R, Dallapiccola B : A syndrome with true anophthalmia, hand-foot defects and mental retardation. Ophthalmic Paediatr Genet 1983; 4: 19–23.
Brunquell PJ, Papale JH, Horton JC et al: Sex-linked hereditary bilateral anophthalmos: pathologic and radiologic correlation. Arch Ophthalmol 1984; 102: 108–113.
Traboulsi EI, Lenz W, Gonzales-Ramos M, Siegel J, Macrae WG, Maumenee IH : The Lenz microphthalmia syndrome. Am J Ophthalmol 1988; 105: 40–45.
Graham CA, Redmond RM, Nevin NC : X-linked clinical anophthalmos: localization of the gene to Xq27-Xq28. Ophthalmic Paediatr Genet 1991; 12: 43–48.
Antoniades K, Tzouvelekis G, Doudou A, Nanas C : A sporadic case of Lenz microphthalmia syndrome. Ann Ophthalmol 1993; 25: 342–345.
Ozkinay FF, Ozkinay C, Yuksel H, Yenigun A, Sapmaz G, Aksu O : A case of Lenz microphthalmia syndrome. J Med Genet 1997; 34: 604–606.
Temtamy SA, Ismail SI, Meguid NA : Lenz microphthalmia syndrome: three additional cases with rare associated anomalies. Genet Couns 2000; 11: 147–152.
Lehman DM, Sponsel WE, Stratton RF et al: Genetic mapping of a novel X-linked recessive colobomatous microphthalmia. Am J Med Genet 2001; 101: 114–119.
Ersin NK, Tugsel Z, Gokce B, Ozpinar B, Eronat N : Lenz microphthalmia syndrome with dental anomalies: a case report. J Dent Child 2003; 70: 262–265.
Gupta A, Srinivasan R, Panadian DG, Babu KR : Lenz microphthalmia syndrome in an Indian patient. Indian J Ophthalmol 2007; 55: 462–463.
Martinez-Garay I, Tomas M, Oltra S et al: A two base pair deletion in the PQBP1 gene is associated with microphthalmia, microcephaly and mental retardation. Eur J Hum Genet 2006; 15: 29–34.
Acknowledgements
EH is funded by the Wellcome Trust (UK) and BDF Newlife (UK). SW is funded by la Fondation pour la Recherche Médicale (France). This research was supported in part by the Intramural Research program of the National Human Genome Research Institute, NIH (US). We thank Patricia Conteville and Nathalie Collot for technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Hilton, E., Johnston, J., Whalen, S. et al. BCOR analysis in patients with OFCD and Lenz microphthalmia syndromes, mental retardation with ocular anomalies, and cardiac laterality defects. Eur J Hum Genet 17, 1325–1335 (2009). https://doi.org/10.1038/ejhg.2009.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.52
Keywords
This article is cited by
-
Oculo-facio-cardio-dental (OFCD) syndrome: a case report
Journal of Medical Case Reports (2024)
-
Oculofaciocardiodental syndrome caused by a novel BCOR variant
Human Genome Variation (2023)
-
A rare genotype of biallelic mosaic variants in BCOR gene causing a bilateral ocular anterior segment dysgenesis and cataracts
European Journal of Human Genetics (2023)
-
Y chromosome is moving out of sex determination shadow
Cell & Bioscience (2022)
-
A novel deletion mutation in the BCOR gene is associated with oculo-facio-cardio-dental syndrome: a case report
BMC Pediatrics (2022)
