
Abstract
Teeth from 38 aboriginal remains of La Palma (Canary Islands) were analyzed for external and endogenous mitochondrial DNA control region sequences and for diagnostic coding positions. Informative sequences were obtained from 30 individuals (78.9%). The majority of lineages (93%) were from West Eurasian origin, being the rest (7%) from sub-Saharan African ascription. The bulk of the aboriginal haplotypes had exact matches in North Africa (70%). However, the indigenous Canarian sub-type U6b1, also detected in La Palma, has not yet been found in North Africa, the cradle of the U6 expansion. The most abundant H1 clade in La Palma, defined by transition 16260, is also very rare in North Africa. This means that the exact region from which the ancestors of the Canarian aborigines came has not yet been sampled or that they have been replaced by later human migrations. The high gene diversity found in La Palma (95.2±2.3), which is one of the farthest islands from the African continent, is of the same level than the previously found in the central island of Tenerife (92.4±2.8). This is against the supposition that the islands were colonized from the continent by island hopping and posterior isolation. On the other hand, the great similarity found between the aboriginal populations of La Palma and Tenerife is against the idea of an island-by-island independent maritime colonization without secondary contacts. Our data better fit to an island model with frequent migrations between islands.
Similar content being viewed by others

Introduction
The Canary Islands are a group of seven Atlantic Islands situated about 60 miles off the northwest African coast. Due to their volcanic origin, they were never a part of the African mainland. However, when the Europeans, along the fourteenth and fifteenth centuries, visited and conquered the archipelago they realized that their inhabitants had a primitive Neolithic culture, no knowledge of seafaring techniques, and very little communication between islands.1 Archeological, anthropological and linguistic studies suggested that the North African Berbers were the most probable ancestors of the Canarian aborigines. This origin was corroborated, at molecular level, when specific North African markers, as mtDNA haplotypes belonging to the U6 subhaplogroup, Y-chromosome haplotypes belonging to the E-M81 subhaplogroup, or the CD4/Alu 110(−) haplotypes were detected in the current Canarian population, attesting the survival of an important aborigine substrate in it.2, 3, 4 Further evidence came from mtDNA analysis carried out directly on historical and aboriginal remains,5, 6 in which U6 lineages were detected at greater frequencies than in the present day Canarian population. However, as no exact matches of the indigenous U6b1 lineages could be found in North Africa,5 it was not possible to establish a precise origin for the pre-Hispanic colonizers. On the other hand, radiocarbon dating7 suggested that, most probably, the archipelago was occupied at the beginning of the first millennium BC. However, it is not clear whether the islands were colonized by one or by several migratory waves. Cultural differences between islands and its evolution within islands could be explained by isolation and indigenous progress. Nevertheless, clear-cut differences in pottery horizons strongly suggested that, at least the eastern islands of Gran Canaria, Fuerteventura and Lanzarote, were secondarily influenced by a more advanced culture.8 Also, for the western island of La Palma, a Saharan cultural influence, dated around the ninth or tenth centuries, has been proposed.9 In any case, it is difficult to distinguish demic from simple cultural influences based only on archeological remains. Genetic studies on diversity and frequency distribution of polymorphic markers in the current populations of the seven islands have also given inconclusive results. The correlation patterns found for mtDNA, CD4/Alu haplotypes, and AB0 allele frequencies were congruent with only one dominant settlement process,2, 3, 10 whereas the distribution and dating of some Y-chromosome North African lineages, present in the Canary Islands, matched with a pattern of settlement of the archipelago in two stages.4 If the aboriginal colonizers reached the islands by themselves, using primitive boating skills, they most probably landed first on the nearest eastern islands from the mainland and, under demographic pressure, ventured afterwards to the next western island at sight. Under this restrictive stepping stone model of colonization, without important secondary migration between islands, a diminution in genetic diversity from the eastern to the western islands is expected in such a way that the islands more distant from the African shore would have only a subset of the variation held by the eastern islands. However, if the aborigines were brought to the islands in several waves by other people with seafaring experience,1 different islands could have been colonized independently, so that their respective genetic diversity would not be correlated with the geographical distance of the islands from the Continent. So, three important issues about the aboriginal colonization of the Canary Islands are still ignored: (a) the precise North African point(s) from which the Canarian ancestors originated, (b) whether the indigenous colonizers reached the islands by themselves or were brought to them by seafaring people, and (c) if there was only one main or several settlements. The study of more aboriginal remains could help to shed light on the first issue if new mtDNA lineages with a precise localization in North Africa were found. Furthermore, as there is an aboriginal sample of the central island of Tenerife previously analyzed for mtDNA,5 the additional analysis of an indigenous sample from one of the two westernmost islands of the Archipelago, El Hierro or La Palma, would make possible their comparison, at the mtDNA gene diversity and structure levels, to test the alternative models of colonization directly on the prehispanic aboriginal population. Due to demographic differences between these two islands at the time of the conquest, with that of El Hierro heavily punished by slavery raids with only around a hundred survivors compared with La Palma ranging from 1200 to over 4000 inhabitants,11 the latter seemed more adequate for the above commented comparisons. With these objectives in mind, in this study, an aboriginal sample from La Palma island was chosen to be analyzed for the hypervariable region I (HVR I) of mtDNA and for the diagnostic coding region positions that allowed the unequivocal assignation of their lineages to one of the known mtDNA haplogroups, so that its relationship with other populations from the islands and North Africa could be accurately established.
Materials and methods
Sample collection
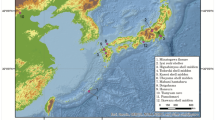
Prehistoric remains were obtained from five different archeological sites excavated in La Palma Island (Figure 1). In all cases, teeth were chosen as the preferable ancient skeletal DNA source. A total of 38 different individuals were sampled and, in five cases, two independent analyses from the same individual were possible (Supplementary Material 1). One sample was from Barlovento, 21 from Puntallana (El Espigón), 9 from Santa Cruz de La Palma, 6 from Mazo, and 1 from Los Llanos de Aridane (Supplementary Material 1). In all cases well preserved teeth were chosen. Although this material was not directly C-14 dated, it is known that the most ancient settlements in La Palma are not older than the first millennium AD.12 Ceramic types co-excavated with teeth in El Espigón site confirmed the relatively recent age of these remains, around 600–1200 AD.

Geographical position of La Palma within the Canary Islands and locations of the archeological sites sampled.
External DNA extraction
To assess whether the teeth were externally contaminated the following protocol was used to extract DNA from their external surfaces: Teeth were repeatedly swabbed using a sterilized swab stick soaked in a guanidinium thiocyanate (GuSCN) washing solution (6 M GuSCN, 0.1 M Tris-HCl, pH 6.4). The swab was introduced in a 1.6 ml tube with 300 μl extracting solution (6 M GuSCN, 0.1 M Tris-HCl, pH 6.4, 0.2 M EDTA, 2% Triton X-100) and the stick broken to close the tube. Then the tube was boiled for 10 min and left to room temperature (RT) for other 10 min. After that, its top and bottom were perforated with a sterile syringe needle and fitted in a new 1.6 ml tube to which the solution was transferred by centrifugation. Fifteen microliters of acid-treated size-fractionated silica particles (Sigma), previously autoclaved, were added and the new tube was kept at RT with occasional vortex during 10 min. Then the silica particles were pelleted by centrifugation, the supernatant discarded and the pellet washed twice with 1:1 washing solution/ethanol, once with acetone and once with 70% ethanol. Finally, the DNA was eluted from the silica pellet resuspending it in 50 μl TE buffer pH 8, incubating for 10 min at 55°C, and transferring supernatant into a new tube after centrifugation to pellet the silica. Five microliters of this solution was used as template for 25 μl volume PCR reactions.
Internal DNA extraction
For internal extraction, teeth were again thoroughly washed, this time, with 15% HCl, rinsed with UV-treated ddH2O, and then exposed to UV light for 10 min. To reconstruct the tooth after extraction, it was cut through the midline using a dentist electric saw and the internal pulp and dentin drilled out using a dental drill. The powder was collected in 1.5 ml sterile tubes. Then DNA was extracted according to a modified GuSCN-silica-based protocol.5
Spectrometric DNA quantification
Ancient DNA (aDNA) was quantified using an ND-1000 (NanoDrop) spectrophotometer following the manufacturer's instructions.
Real-time PCR quantification
Real-time PCR quantification was carried out to assess the number of molecules used as template for PCR amplification.13 We used iQ SYBR Green Supermix (BioRad) in a iCycler Thermal Cycler (BioRad). Primers and thermal cycling conditions were as previously specified.5 Tenfold serial dilutions of a purified and quantified standard were included in the experiment to determine the standard curve, to estimate the initial number of DNA molecules in each sample.
Amplification
For mtDNA HVRI sequencing or RFLP analysis on aborigine samples, primers pairs and PCR conditions used were as previously published.5, 14 To improve the reading of each HVRI fragment sequence, the M13 forward primer (Promega) was added, as a tail, at the 5′ end of all the seven forward primers used. In addition, two new RFLP analyses, that characterise subhaplogroups H115 and H3,16 were performed. The first was typed amplifying a fragment of 50 bp, using the forward primer L3005 5′-CTCGATGTTGGATCAGGACA-3′ and a modified reverse primer H3011 5′-GAACCTTTAATAGCGGCTGCACAAT-3′ that creates a Tsp509I site when the derived A is present, giving fragments of 24 and 26 bp. The second was checked amplifying a fragment of 53 bp, using the forward primer L6769 5′-CTAGGGTTTATCGTGTGAGC-3′ and a modified reverse primer H6777* 5′-TCTACGTCTATTCCTACTGTACATAT-3′ that creates an NdeI site when the derived C is present, giving fragments of 25 and 28 bp.
For HVRI amplification of the external DNA extracts, primers HVI (L15840) and HVII (H16436) and conditions as detailed elsewhere17 were used. In case this long-fragment amplification failed, fragments of approximately half size were amplified using primer pairs HVI/H3R and L3F/HVII.5 Complete mtDNA amplification, in present day samples, was carried out after all the aDNA analyses were completed. Primer pairs and conditions were as described earlier.18, 19
Cloning
Amplification products were cloned into pGEM-T vectors (Promega). Several clones were sequenced for each fragment until an unambiguous consensus sequence was obtained. Additional cloning was carried out in those cases in which only one of the three negative PCR controls showed contamination, and this was of a much lower intensity than the sample amplification. Cloned sequences from the sample were used only when: (a) detected variants were different between the contaminated control and the aboriginal sample, and (b) the mutations observed for that segment were phylogenetically congruent with the haplotype obtained with the rest of the fragments.
Sequencing
Ancient DNA PCR fragments were directly sequenced with the M13 forward primer. Clones were sequenced using the M13 reverse primer (Promega). Amplified fragments from external DNA extracts and from modern samples were sequenced with the same forward primer used in the PCR reaction. When necessary the reverse strand was also sequenced. All the sequencing reactions were prepared with the BigDye v3.1 Terminator Cycle Sequencing kit (Applied Biosystems) using 10 μl volume reactions. At La Laguna, sequencing reactions were run on a MegaBACE 1000 sequencing system (Amersham Pharmacia Biotech). At Las Palmas they were run on an ABI PRISM 310 Genetic Analyzer (Applied Biosystems). To confirm the accuracy of the fragments amplified for RFLP diagnosis, half of their PCR reactions were directly sequenced at least in two samples. DNA sequences were analyzed using CLUSTAL X 1.83 and confirmed by inspecting the electropherograms manually.
Criteria of authenticity
Standard contamination prevention and monitoring used at La Laguna and at Las Palmas laboratories were as detailed earlier.5, 14 We tried to follow the eight criteria by Pääbo et al.,20 which include those set forth in Cooper and Poinar.21
-
1
Amplification products have been cloned, multiple clones sequenced, and misincorporated bases detected in several instances.
-
2
One blank extraction and three negative PCR controls per sample and amplification have been always performed.
-
3
Consistence of the sequences obtained has been confirmed by repeated amplification and by RFLP analysis that, in all cases, matched the haplogroup assignation based on HVSI sequencing.
-
4
The initial number of amplifiable DNA molecules has been quantified using real-time PCR.
-
5
To facilitate the aDNA amplification, we analyzed the mtDNA HVSI region using seven overlapping small fragments. Although in some instances, we were able to obtain double fragments, the total HVSI region could not be amplified in any case.
-
6
Due to the lack of the appropriate technology, biochemical assays of macromolecular preservation could not be performed.
-
7
Our overlapping approach would detect any amplification of mtDNA sequences integrated into the nuclear genome.
-
8
Five duplicated samples were assayed in a second independent laboratory.
-
9
Although there were no associated faunal remains to be analyzed, we had previously extracted mtDNA from lizard remains, obtained from caves with similar environmental conditions as those for the aboriginal graves.22 Moreover, it has to be taken into account that the estimated age for these animal remains was older than the oldest date for La Palma aboriginal samples.
With the aim to remove and to detect any previous surface modern DNA contamination, two additional criteria have been followed:
We have previously tested that the decontamination step in our extraction protocol was stringent enough to remove chicken DNA contamination put on the surface of teeth.5
In addition, in this study, any modern DNA contamination present on the surface of the teeth was retrieved before the extraction protocol for endogenous DNA was carried out. This approach allowed the subsequent comparison of both DNA sources.
Statistical analysis
Sequences were sorted into haplogroups following the most actualized classification,23 further updated for J1b.19 Gene diversity was calculated according to Nei.24 The published samples, used for comparisons, are detailed in Supplementary Material 2. The relationships between populations were estimated using haplogroup and haplotype frequency-based linearized FST, computed by the Arlequin 2000 program.25 Both data sets were used to obtain multidimensional scaling plots by means of the SPSS ver 13.0 package (SPSS Inc., Chicago, Illinois). In addition, the absolute number of migrants exchanged between the two aboriginal populations (M) was also estimated using FST based on haplotype frequencies (Arlequin 2000 program). To diminish the strong influence of the common haplotypes in FST based distances, an additional measure of haplotypic identity (IXY=(HTXY2/(HTX HTY)) was used, where HTXY is the number of shared haplotypes between populations X and Y, and HTX and HTY are the number of different haplotypes in populations X and Y, respectively. Phylogenetic relationships among HVSI and genomic mtDNA sequences were established using the reduced median network algorithm.26 Coalescence ages, based on HVSI sequences, were estimated using the ρ-statistic27 and a calibration of 1 transition per 20 180 years.28
Results
External extracts for all the 38 samples available for the molecular analysis were obtained. For 21 teeth, no surface sequences could be retrieved. In three cases, the external extract was contaminated by the researcher in charge of the analysis (VMC). For the remaining 18 ones, the external sequences obtained belonged to the two anthropologists who previously performed physical measures on the teeth. In two cases, after cloning, both anthropologist sequences could be identified on the same tooth (Supplementary Material 1). However, the sequence from the archeologist, who participated in all the excavations and in the conservation of this material, was not retrieved in any case. Real-time PCR quantification gave a 3726±2400 mean number of molecules used as initial template, being 931 molecules the minimal estimation. Endogenous informative mtDNA sequences were obtained from a total of 30 individuals, accounting for an efficiency of 78.9%. It seems that poor DNA content and the presence of inhibitors were the main causes for the lack of amplification in the remaining eight samples, as they gave spectrophotometric lower DNA sample concentration (4.7±0.8 ng/μl) and anomalous 260/280 nm absorbance ratios (3.3±1.6) compared with the successful group, 11.5±0.4 and 1.7±0.4, respectively. Inhibition was also a problem for the real-time PCR quantitative molecular estimation as in several cases, samples had to be diluted to get optimal amplification. Cloning was carried out on 15 samples (Supplementary Material 1) and a mean of five clones were sequenced by sample. They were chosen because they had slight PCR contamination in one of the fragments or because, although no contamination was detected, their haplotypes shared some mutations with some of the researchers involved in the analysis. In all cases, the consensus cloned sequences were the same as the corresponding direct sequences. Moreover, the contaminated fragment sequence could be always distinguished from the endogenous haplotype. In addition, the following mutations (16085, 16175, 16176G, 16222, 16235, 16256, 16287, 16298, 16299, 16318T, 16332, 16343, 16354, 16364, and 16388) appeared individually in some of the clones. Because of its sporadic detection and lack of phylogenetic sense within the consensus sequence obtained, its presence was attributed to postmortem DNA damage. Due to its very little phylogenetic information, fragments 1 and 2 were analyzed only when enough DNA extract remained after the other five fragments and the RFLP analysis were carried out. The five samples replicated in Las Palmas resulted identical to those obtained in La Laguna (Supplementary Material 1). In some cases, aboriginal haplotypes shared one or two positions with some of the researchers involved in the molecular analysis. However, in all cases, the haplogroup ascription of these haplotypes was different. For instance, the aborigine sample PA-17 shares transition 16316 with VMC but, whereas the latter belonged to subgroup H1, the former did not (Table 2). In addition, PA-17 lacked 16129 transition present in the VMC haplotype. Furthermore, no external contamination was obtained from this sample (Supplementary Material 1).
Table 1 compares haplogroup frequencies between aboriginal, historical, and modern samples from the Canary Islands and those from Northwest (Morocco, Mauritania, and Sahara) and North Central Africa (Algeria and Tunisia). In general, haplogroup frequencies in aborigines from La Palma and Tenerife are highly coincident (FST=0.0047, P=0.30), although some haplogroups present in La Palma, such as T1a, W and X were not detected in Tenerife and haplogroup HV0, and subgroup U6a1/a2/a3, present in Tenerife, were not found in La Palma. This could be attributed to small sample size. Curiously, the U6a1/a2/a3 type was also absent in the modern population of La Palma. By far, haplogroup H was the most abundant in the aborigines from La Palma (56.7%) and Tenerife (47.2%). Although there are no significant differences with the historical sample from Tenerife, nor with the modern population of the Archipelago, there was a significant excess of H lineages in the aboriginal sample when compared with North Africa (P<0.005). Macro-haplogroup L was also present in moderate frequencies in the aboriginal populations of La Palma (6.7%) and Tenerife (11.1%). However, as for H, there was only significant differences (P=0.003) with L frequencies (24.8%) in North Africa.
Haplotypic frequencies and RFLP analyses performed in the aboriginal population of La Palma are presented in Table 2. Twenty different haplotypes (67%) were found in the 30 individuals analyzed giving a gene diversity of 95.2±2.3 that was not significantly different from the Tenerife aboriginal sample (92.4±2.8). Although, a slight level of endogamic isolation among aboriginal groups within La Palma and within Tenerife could be deduced from the high frequency of the 16260 haplotype in the Espigón (La Palma) and the CRS type in CP (Tenerife) (Supplementary Material 3), both aboriginal samples presented diversity values not significantly different from their respective modern populations (Table 2). The majority (14) of the aboriginal haplotypes from La Palma (70 %) had exact matches in North Africa (Table 2). Matches for the six remaining ones were of different ascription. The Canarian U6b1 ancestral haplotype (16163 16172 16219 16311) has only exact matches within the Archipelago. Its occasional presence in Spain and South America has been attributed to the historical Canarian emigration.29, 30 Of the two haplotypes, which are one-step derived from the most abundant H type of the aboriginal sample (16260), one (16192 16260) belonging to the H3 subgroup has only been found in the modern population of La Palma. The other one (16260 16278) of H1 ascription has never been detected before because, although the same HVRI motif was found in a Yemeni sample,31 it belonged to H14 subgroup. Finally, the other 3 (15%) haplotypes have only been found in the islands and in the European side of the Mediterranean basin. The aboriginal 16260 H1 type deserved special attention because, within haplogroup H, this transition is very redundant as it has been independently observed in at least 11 different subgroups as deduced from the whole sequencing of six 16260 lineages and the unequivocal subhaplogroup H assignation of eight additional partial sequences (Figure 2). So, RFLP analyses have to be performed to accurately establish exact haplotypic matches. All but one aboriginal 16260 haplotype belonged to the H1 subgroup (Table 2), however, for the modern populations of the Archipelago, albeit H1 is also the most frequent, there are also H3 and H7 representatives (Figure 2). In North Africa, the H1 16260 haplotype has only been detected once in North Central Africa as that found in Northwest Africa, belonged to the H3 subgroup. Furthermore, contrarily to the Canary Islands, 16260 H1-derived haplotypes have not yet been found in North Africa. Due to the relative recent radiocarbon dates estimated for the aboriginal settlement of the Archipelago,7 it seems improbable that all the derived 16260 types were originated in the islands after their colonization. Their absence or rareness in North Africa, likewise that for the U6b1 motif, has to be explained because the precise site from which the Canarian ancestors originated in North Africa has not yet been sampled or because they have been replaced by later human migrations.29 In fact, ρ-values for both 16260 H1 and U6b1 authoctonous clusters gave coalescence ages of 6263±2869 and 5911±3536 years ago, respectively. These dates, not statistically different, clearly precede the age of the indigenous colonization, proposed by radiocarbon dates and archeological criteria at the beginning of the first millennium. Attending to global exact matches between samples, the highest relationship of aborigines from La Palma was with aborigines from Tenerife (0.165) and the most distant populations were those from Northwest (0.018) and North Central (0.016) Africa. Among islands, the aboriginal sample from La Palma had greater affinities with the western islands (0.095±0.020), to which it geographically belongs, than with the eastern islands (0.051±0.003). The high number of migrants exchanged between the two aboriginal population of La Palma and Tenerife (M=103), reinforces the high relationship found at haplogroup and haplotype levels.

Phylogenetic tree of complete (continuous branches) and incomplete (discontinuous branches) haplogroup H mtDNA sequences carrying the 16260 transition. Numbers along links refer to nucleotide transitions. ‘d’ Indicate deletions and ‘i’ insertions. Recurrent mutations are underlined. Sequence references: CRS;35, 36 SYR456 and YEM1615;31 Can196, CanA07, CanD295, CanD508, Can756, and Alg6402 (this study; GenBank accession nos. J719302-FJ719307); AUT5F8, AUTZa9, AUTf1F5, and AUT3F9;37 GER54;38 POR162.39 Geographic origins: Alg6402: Algerian; AUT5F8, AUTZa9, AUTf1F5, and AUT3F9: Austrians; Can196, CanA07, CanD295, CanD508, and Can756: Canarian; GER54: German; POR162: Portuguese; SYR456: Syrian; YEM1615: Yemeni.
The relationships among populations based on haplogroup and haplotype FST distances are graphically shown in Figure 3. In both cases, La Gomera was the most divergent population mainly due to its comparatively low frequency of haplogroup H (21.7%) and because it presents the highest frequency of the Canarian indigenous U6b1 motif (47.8%). It is also worth mentioning that the aboriginal population from La Palma was always nearer to the aboriginal population from Tenerife than to the modern population of La Palma, whereas the aborigines from Tenerife are closer to their present day descendants in the island. Attending to haplogroup frequencies, Gran Canaria was the nearest island to North Africa. However, when haplotypic frequencies were used, the historical sample from Tenerife was the closest. This apparent discrepancy was due to the fact that exact L haplotypic matches between North Africa and the historical sample from Tenerife 5 were greater than between the former and Gran Canaria.2

Graphical relationships among the studied populations. MDS plots based on FST haplotypic (a) and haplogroup (b) frequency distances. Codes are as in Table 1.
Discussion
Before any conclusion based on aDNA could be established, the authenticity of the data obtained should be assessed. Although the repetition of the results is not a warranty of lack of modern DNA contamination,20 the fact that modern contaminating DNA was retrieved from the surface of about 50% of the teeth, and that the endogenous sequences obtained did not match the external ones, seems to be an additional criterion for authenticity. It has been demonstrated that the ancient remains are most susceptible to contamination at the excavation process32 and subsequent museum preservation.33 However, from our results, it seems that well preserved teeth are less sensible to these routes of contamination. In our case, the archeologist sequence was not retrieved from the external extracts, possibly due to its elimination by later anthropologists handling. Furthermore, this sequence did not appear as a contaminant of the endogenous extracts. In addition, no more than one sequence, with phylogenetic sense, could be constructed from the mutations consistently detected in the different PCR fragments and clones of a same sample. Therefore, we feel confident of the reliability of our results.
On the basis of the probabilistic criteria, eight sequences of putative Northwest African ancestry were proposed as Canarian pre-Hispanic founder haplotypes and 13 additional ones, derived from them, as possible additional founder types.2 Five of these basic founders were detected in the aboriginal sample of La Palma, where only the U6c1 type was absent.29 However, previous5 and the present aDNA study on prehistoric remains have shown that the number of founder lineages was higher than expected. In total, 36 additional lineages have been found. The bulk of them26 belonged to West Eurasian haplogroups, being the rest10 of sub-Saharan Africa ascription. As expected from archeological, anthropological and linguistic studies, the majority of the West Eurasian and sub-Saharan African lineages had exact matches in North Africa. Furthermore, there is an indigenous North African haplogroup, U6,34 that has Canarian aboriginal representatives in its three main branches (U6a, U6b, U6c), which strongly points to this part of Africa as the most probable origin for the pre-Hispanic Canarian colonizers. However, the fact that the specific Canarian subtype U6b1 has not yet been found in North Africa makes the assignation of a precise geographic origin difficult. In addition, it has to be taken into account that there is a significant portion of aboriginal lineages (15%) that have specific matches to the European side of the Mediterranean basin or to the near East and that, until now, have not been found in North Africa. So, a minor demic component from these areas, on the maternal structure of the pre-Hispanic Canarians, cannot be discarded. This possibility introduces the question of the way(s) and time(s) the islands were colonized before the Spanish conquest. The great gene diversity found in the aborigines from La Palma (95.2±2.3), one of the westernmost islands, similar to that of the aborigines from the central Tenerife Island (92.4±2.8), was an unexpected result. It was also surprising that the two aboriginal samples showed diversities in the same range as the current insular population (Table 2). As samples in each island were collected from four different sites, although with a predominant one (Espigón in La Palma 18 of 30 and CP in Tenerife 24 of 36), we wondered whether population structure within islands existed and the sampling process introduced a bias augmenting diversity. To test this possibility, H was calculated only for the most abundant sites. However, their values were even higher than those obtained for the total sample (96.1±3.0 in Espigón and 93.1±3.2 in CP). So we discarded the existence of a strong population structure and considered the high H values real. These results are against the supposition of a settlement process through island hopping and posterior isolation. Therefore, a restrictive stepping stone model would not fit with the maternal genetic pattern found, although it could be accepted supposing frequent secondary migrations between islands. Under a neutral model, the most important evolutionary forces that modulate gene variation are mutation, recombination, migration, and genetic drift. In the case of mtDNA, recombination is absent. On the other hand, the relative recent age of the Canary Island colonization, suggests that mutation could play only a minor role. In fact, coalescence ages of around 6000 years ago found for two founder clades, pointed to an arrival of both, ancestral and derived lineages, from the North African source population. This leaves migration and genetic drift as the two main counteracting factors to explain the high gene diversity present in the islands, as well as, its genetic resemblance. As commented before, the most accepted age for the aboriginal colonization of the island is at the beginning of the first millennium, and the mean age for the analyzed aboriginal remains is around 900 AD. So, assuming a generation time of 25 years, 36 generations passed since the initial colonization of the two islands. During this period, a total of 103 migrants was estimated, which roughly gives an interchange of around three migrants per generation. This value is sufficient to homogenize their genetic pools and would explain the great similarity found between both populations. However, this migratory input could be an overestimation if, as has been suggested, the colonizers of the two islands had the same North African origin.
Under these assumptions, an island-by-island independent maritime colonization, without secondary migrations, is also improbable. Moreover, if the Canary Islands were colonized in several waves, most probably Tenerife and La Palma were affected likewise by these events. At present, our data better fit to an island model with frequent migrations between islands. Future studies on aboriginal remains from other islands and from modern and ancient North African populations would help to elucidate these remaining questions.
Accession codes
References
Mercer J : The Canary Islanders: their prehistory, conquest, and survival. London, Rex Collings 1980.
Rando JC, Cabrera VM, Larruga JM et al: Phylogeographic patterns of mtDNA reflecting the colonization of the Canary Islands. Ann Hum Genet 1999; 63: 413–428.
Flores C, Maca-Meyer N, Pérez JA, Cabrera VM : The peopling of the Canary Islands: a CD4/Alu microsatellite haplotype perspective. Hum Immunol 2001; 62: 949–953.
Flores C, Maca-Meyer N, Pérez JA, González AM, Larruga JM, Cabrera VM : A predominant European ancestry of paternal lineages from Canary Islands. Ann Hum Genet 2003; 67: 138–152.
Maca-Meyer N, Arnay M, Rando JC et al: Ancient mtDNA analisis and the origin of the Guanches. Eur J Hum Genet 2004; 12: 155–162.
Maca-Meyer N, Cabrera VM, Arnay M et al: Mitochondrial DNA diversity in 17th–18th century remains from Tenerife (Canary Islands). Am J Phys Anthropol 2005; 127: 418–426.
Delgado-Darias T, Velasco-Vazquez J, Arnay de la Rosa M, Martin Rodriguez E, Gonzalez-Reimers E : Dental caries among the pre-Hispanic population from Gran Canaria. Am J Phys Anthropol 2005; 128: 560–568.
Abercromby J : Prehistoric pottery of the Canary Islands and its makers. J Royal Anthropol Inst of Great Britain and Ireland 1914; 44: 302–323.
Navarro Mederos JF : Pasado y presente de la arqueología canaria. Espacio, Tiempo y Forma. Prehistoria y Arqueología Canaria 1997; 10: 447–478.
Fregel R, Maca-Meyer N, Cabrera VM, González AM, Larruga JM : Description of a simple multiplex PCR-SSCP method for AB0 genotyping and its application to the peopling of the Canary Islands. Immunogenet 2005; 57: 572–578.
Macias-Hernández A : Fuentes y principales problemas metodológicos de la demografía histórica de Canarias. Anuario de Estudios Atlánticos 1988; 34: 51–158.
Martín E : La Palma y los auaritas. Santa Cruz de Tenerife, Centro de la Cultura Popular Canaria 1992.
Alonso A, Martín P, Albarrán C et al: Real-time PCR designs to estimate nuclear and mitochondrial DNA copy number in forensic and ancient DNA studies. For Sci Int 2003; 139: 141–149.
Casas MJ, Hagelberg E, Fregel R, Larruga JM, González AM : Human mitochondrial DNA diversity in an archaeological site in al-Andalus: genetic impact of migrations from North Africa in Medieval Spain. Am J Phys Anthropol 2006; 131: 539–551.
Finnilä S, Lehtonen MS, Majamaa K : Phylogenetic network for European mtDNA. Am J Hum Genet 2001; 68: 1475–1484.
Herrnstadt C, Elson JL, Fahy E et al: Reduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian and European haplogroups. Am J Hum Genet 2002; 70: 1152–1171.
González AM, Cabrera VM, Larruga JM et al: Mitochondrial DNA variation Mauritania and Mali and their genetic relationship to other Western Africa populations. Ann Hum Genet 2006; 70: 631–657.
Maca-Meyer N, González AM, Larruga JM, Flores C, Cabrera VM : Major genomic mitochondrial lineages delineate early human expansions. BMC Genetics 2001; 2: 13–20.
Abu-Amero KK, Larruga JM, Cabrera VM, González AM : Mitochondrial DNA structure in the Arabian Peninsula. BMC Evol Biol 2008; 8: 45.
Pääbo S, Poinar H, Serre D et al: Genetic analyses from ancient DNA. Annu Rev Genet 2004; 38: 645–679.
Cooper A, Poinar HN : Ancient DNA: do it right or not at all. Science 2000; 18: 289–1139.
Maca-Meyer N, Carranza S, Rando JC, Arnold EN, Cabrera VM : Status and relationships of the extinct giant Canary Island lizard Gallotia goliath (Reptilia: Lacertidae), assessed using ancient mtDNA from its mummified remains. Biol J Linn Soc 2003; 80: 659–670.
van Oven M, Kayser M : Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 2008; online.
Nei M : Molecular evolutionary genetics. New York: Columbia University Press, 1987.
Schneider S, Roessli D, Excoffier L : Arlequin ver. 2.0: A software for population genetic data analysis. Genetics and Biometry Laboratory. Switzerland: University of Geneva, 2000.
Bandelt H-J, Forster P, Röhl A : Median-ing networks for inferring intraspecific phylogenies. Mol Biol Evol 1999; 16: 37–48.
Saillard J, Forster P, Lynnerup N, Bandelt HJ, Norby S : mtDNA variation among Greenland Eskimos. The edge of the Beringian expansion. Am J Hum Genet 2000; 67: 718–726.
Forster P, Harding R, Torroni A, Bandelt H-J : Origin and evolution of Native American mtDNA variation. A reappraisal. Am J Hum Genet 1996; 59: 935–945.
Maca-Meyer N, González AM, Pestano J, Flores C, Larruga JM, Cabrera VM : Mitochondrial DNA transit between West Asia and North Africa inferred from U6 phylogeography. BMC Genetics 2003; 4: 15.
Plaza S, Calafell F, Helal A et al: Joining the pillars of Hercules. mtDNA sequences show multidirectional gene flow in the western Mediterranean. Ann Hum Genet 2003; 67: 312–328.
Roostalu U, Kutuev I, Loogvali E-L et al: Origin and expansion of haplogroup H, the dominant human mitochondrial DNA lineage in West Eurasia: the Near East and Caucasian perspective. Mol Biol Evol 2007; 24: 436–448.
Sampietro ML, Gilbert MTP, Lao O et al: Tracking down human contamination in ancient human teeth. Mol Biol Evol 2006; 23: 1801–1807.
Pruvost M, Schwarz R, Correia VB et al: Freshly excavated fossil bones are best for amplification of ancient DNA. Proc Nat Acad Sci USA 2007; 104: 739–744.
Rando JC, Pinto F, González AM et al: Mitochondrial DNA análisis of northwest African populations reveals genetic exchanges with European, Near Eastern and sub-Saharan populations. Ann Hum Genet 1998; 62: 531–550.
Anderson S, Bankier AT, Barrell BG et al: Sequence and organization of the human mitochondrial genome. Nature 1981; 290: 457–465.
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N : Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 1999; 23: 147.
Brandstaetter A, Zimmermann B, Wagner J et al: Timing and deciphering mitochondrial DNA macrohaplogroup R0 variability in Central Europe and Middle East. BMC Evol Biol 2008; 8: 191.
Poetsch M, Witting H, Krause D, Lignitz E : Mitochondrial diversity of a northeast German population. For Sci Int 2003; 137: 125–132.
Pereira L, Richards M, Goios A et al: Evaluating the forensic informativeness of mtDNA haplogroup H sub-typing on a Eurasian scale. Foren Sci Int 2006; 159: 43–50.
Acknowledgements
We gratefully acknowledge P Hernández Carpio and A González and J Espino from SEGAI for technical assistance and to N Maca-Meyer and A Gámez for their helpful comments. The samples were generously donated by J Pais. This research was supported by grants from the Spain Ministry of Science and Technology BFU2006-04490 to JM Larruga. R Fregel was supported by a Gobierno de Canarias predoctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Rights and permissions
About this article
Cite this article
Fregel, R., Pestano, J., Arnay, M. et al. The maternal aborigine colonization of La Palma (Canary Islands). Eur J Hum Genet 17, 1314–1324 (2009). https://doi.org/10.1038/ejhg.2009.46
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.46
Keywords
This article is cited by
-
The genomic history of the indigenous people of the Canary Islands
Nature Communications (2023)
-
An Evolutionary Approach to the History of Barley (Hordeum vulgare) Cultivation in the Canary Islands
African Archaeological Review (2020)
-
Morphological and genetic characterization of barley (Hordeum vulgare L.) landraces in the Canary Islands
Genetic Resources and Crop Evolution (2019)
-
Population structure of eleven Spanish ovine breeds and detection of selective sweeps with BayeScan and hapFLK
Scientific Reports (2016)
-
Detecting the existence of gene flow between Spanish and North African goats through a coalescent approach
Scientific Reports (2016)
