
Abstract
Focal dermal hypoplasia (FDH) is an X-linked developmental disorder with male lethality characterized by patchy dermal hypoplasia, skeletal and dental malformations, and microphthalmia or anophthalmia. Recently, heterozygous loss-of-function mutations in the PORCN gene have been described to cause FDH. FDH shows some clinical overlap with the microphthalmia with linear skin defects (MLS) syndrome, another X-linked male lethal condition, associated with mutations of HCCS in the majority of cases. We performed DNA sequencing of PORCN in 13 female patients with the clinical diagnosis of FDH as well as four female patients with MLS syndrome and no mutation in HCCS. We identified PORCN mutations in all female patients with FDH. Eleven patients seem to have constitutional PORCN alterations in the heterozygous state and two individuals are mosaic for the heterozygous sequence change in PORCN. No PORCN mutation was identified in the MLS-affected patients, providing further evidence that FDH and MLS do not overlap genetically. X chromosome inactivation (XCI) analysis revealed a random or slightly skewed XCI pattern in leukocytes of individuals with intragenic PORCN mutation suggesting that defective PORCN does not lead to selective growth disadvantage, at least in leukocytes. We conclude that the PORCN mutation detection rate is high in individuals with a clear-cut FDH phenotype and somatic mosaicism can be present in a significant proportion of patients with mild or classic FDH.
Similar content being viewed by others


Introduction
Focal dermal hypoplasia (FDH), also known as Goltz–Gorlin syndrome (MIM no. 305600), is a rare congenital ectomesodermal disorder characterized by a combination of skin defects, skeletal abnormalities, and ocular anomalies.1 Affected individuals have patchy dermal hypoplasia, often in a distribution pattern following the Blaschko's lines, and areas of subcutaneous fat herniation or deposition of fat into the dermis. In addition, sparse and brittle hair, hypoplastic nails and papillomas have been described. Skeletal abnormalities usually comprise syndactyly, ectrodactyly, and brachydactyly, and in some cases osteopathia striata has been seen. Patients with FDH frequently have ocular anomalies, including microphthalmia/anophthalmia, coloboma, and pigmentary and vascularization defects of the retina. Dental abnormalities, such as hypodontia, abnormal spacing of hypoplastic, or malformed teeth and enamel defects are often present.2 FDH shows phenotypic overlap with the microphthalmia with linear skin defects (MLS) syndrome (MIM no. 309801). The most consistent features of MLS syndrome are microphthalmia with cloudy cornea or sclerocornea and linear skin defects restricted to the face and neck. Some patients also have agenesis of the corpus callosum, chorioretinal abnormalities, infantile seizures, congenital heart defect, and diaphragmatic hernia. Both FDH and MLS syndrome belong to the group of X-linked disorders with male lethality or reduced survival of affected males.3 In the early 1990s it has been hypothesized that FDH and MLS syndrome as well as Aicardi syndrome (MIM no. 304050), another X-linked condition characterized by CNS and eye abnormalities that mainly affects female patients are allelic disorders and represent variable outcomes of the same or a related genetic defect.4, 5, 6 Indeed, the three disorders are characterized by similar anomalies of the brain (agenesis of the corpus callosum), eye (microphthalmia, coloboma, and retinal pigmentary defects), and skin (linear skin defects) that has led to the hypothesis that the genes for these syndromes might be contiguous on the X chromosome.4, 7 However, other authors were rebutting this concept and stated that MLS syndrome should be distinguished from FDH as marked clinical differences exist between the two disorders. For example, skin lesions are limited to the face and neck in MLS syndrome and skeletal abnormalities, such as clefting of hands or feet, and syndactyly are only present in patients with FDH.8, 9
Recently, the underlying genetic defects of FDH and MLS syndrome have been identified. Heterozygous mutations in the HCCS gene (Xp22.2), encoding the mitochondrial holocytochrome c-type synthase are responsible for MLS.10 In patients with FDH, heterozygous mutations in PORCN, located in Xp11.23, have been detected.11, 12 Since the identification of PORCN as the gene responsible for FDH only four additional cases with novel intragenic PORCN alterations have been reported.13, 14 PORCN codes for a member of the Porcupine protein family which are membrane-bound endoplasmatic reticulum proteins. Porcupine proteins are thought to be required for the attachment of long-chain fatty acids to Wnt proteins that is important for proper secretion of Wnt.15, 16 The identification of different genes for FDH and MLS has provided proof that the hypothesis of allelism was erroneous from the beginning. However, not all patients with clinical features highly suggestive of MLS and apparently normal karyotype have a detectable mutation in HCCS (K. Kutsche, unpublished data and Morleo et al17). This finding raises the question whether patients clinically diagnosed with MLS might in rare cases have a PORCN mutation due to clinical and/or genetic overlap between FDH and MLS.
Here, we present results of PORCN mutation screening in 13 female patients with FDH and four female patients displaying clinical manifestations compatible with the diagnosis of MLS syndrome, who had been previously tested negative for a mutation in HCCS.
Patients and methods
Subjects
We collected blood samples or received DNA from 13 female patients clinically diagnosed with FDH and four female patients with MLS syndrome (MLS1-4), who were assessed by experienced clinicians. Clinical information pertinent to the diagnosis was provided by the referring physicians. Table 1 summarizes the clinical findings of individuals with FDH.
MLS1 is a 2½-year-old girl and the first child of healthy parents. She was born at 43 weeks of gestation by natural delivery. The proposita's birth weight was 3570 g (10–50th centile), her body length 51 cm (10–50th centile), and she had an OFC of 34.5 cm (10th centile). At birth, linear erythematous lesions of the skin located on her face, ear lobes, and her left index finger were noticed. Sonography revealed absent corpus callosum and dilated ventricles. Seizures began at the age of 5 months. At 6 months of age, she underwent heart transplantation because of severe dilatative cardiomyopathy. At 15.5 months, her height and weight were below the 3rd centile, and she was microcephalic (OFC 41 cm, −4.6 SD). Dentition was markedly delayed. Ophthalmologic examination revealed myopia on the right eye as well as bilateral strabismus and nystagmus. Muscle tone was low at this age, and the girl was not able to sit without support.
Detailed clinical findings of a female patient MLS2 have previously been described.18 Briefly, she presented with some facial dysmorphic signs and showed linear cutaneous aplasia on her face and neck. Microphthalmia has also been noticed. Cardiac abnormalities included cardiomyopathy in terms of non-compaction myocard accompanied by Wolff–Parkinson–White syndrome. Her psychomotor development was normal.
Patient MLS3, a 5-year-old girl, is the first child of healthy parents. At 41 weeks of gestation, delivery was performed by caesarean section because of decelerating fetal heart rate. Body weight (2600 g) and length (45 cm) were below the 3rd centile, and OFC (32.5 cm) was at the 3rd centile. She presented with multiple linear necrotic skin lesions on her face and neck along the lines of Blaschko. Moreover, the patient had a diaphragmatic hernia, renal agenesis on the left and an L-shaped (tandem) kidney on the right. Sonography of the brain disclosed a small corpus callosum. Echocardiographic analysis demonstrated hypertrophic cardiomyopathy and pulmonary hypertension. At age 15.5 months, she was microcephalic (OFC 42.5 cm, −3.4 SD) and showed the following dysmorphic facial signs: mild facial asymmetry with telecanthus and bilateral epicanthus, broad nasal bridge, smooth philtrum, small mandible, and mildly low-set and protruding ears with hypoplastic helices. Ophthalmologic evaluation revealed pigmentary defects of the retina as the only ocular abnormality. Other physical findings included lack of the 2nd incisor of the left maxilla, talipes on the right, and pes adductus on the left. The patient showed psychomotor retardation.
Only very limited clinical information was available for patient MLS4. MLS4 is a 2-year-old girl who presented with a skin defect on her forehead, microphthalmia/anophthalmia, and an atypical facial cleft.
By routine chromosome analysis, a normal female karyotype with 46,XX was identified in the four patients with MLS syndrome. These individuals had previously been tested negative for mutations in HCCS (PCR and sequencing as well as copy number analysis by quantitative PCR; data not shown). Analysis of the X chromosome inactivation (XCI) status using the androgen receptor assay revealed severely skewed XCI in all four patients with a pattern ranging from 96:4 to 100:0 (K. Kutsche, unpublished data) that is in line with extensive XCI skewing in patients with MLS and HCCS mutation.10, 19
Ethical approval for this study was obtained and informed consent for the genetic analyses was received from parents or their legal guardians.
Mutation and SNP analysis
We isolated genomic DNA from blood lymphocytes of the patients and parents by standard procedures. We amplified the coding region including the flanking intronic sequences of the PORCN gene (14 exons; GenBank accession no. NM_203475) from genomic DNA. Primer sequences are available on request. Amplicons were directly sequenced using the ABI BigDye Terminator Sequencing Kit (Applied Biosystems, Weiterstadt, Germany) and an automated capillary sequencer (ABI 3130; Applied Biosystems). Where mutations were shown to have arisen de novo, we verified declared relationships by genotyping of both parents and the patient at 10 microsatellite loci.
For FDH4 and 7, the PCR product with the mutation was cloned in the pCR2.1 TOPO TA Cloning Vector (Invitrogen, Karlsruhe, Germany). Individual E.coli clones were picked and subjected to colony PCR using standard protocols. PCR products were analyzed by the single-strand conformational polymorphism (SSCP) method. Individual fragments that were assigned to either of the two different banding patterns were sequenced.
Calculation of splicing efficiencies
Splicing efficiencies of the normal versus the newly introduced splice acceptor site in intron 4 of PORCN were calculated by use of three splice prediction programs: SpliceView (http://bioinfo.itb.cnr.it/oriel/splice-view.html), the Berkely Drosophila Genome Project (BDGP) (http://www.fruitfly.org), and NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/).
PORCN transcript analysis
Fresh venous blood samples (2.5 ml) were collected into PAXgene Blood RNA Tubes (PreAnalytiX; QIAGEN, Hilden, Germany). Total RNA was extracted by use of the PAXgene Blood RNA Kit (QIAGEN) according to the manufacturer's instructions. 1 μg of RNA was reverse transcribed into cDNA (Superscript II; Invitrogen) using random hexanucleotides (Invitrogen) according to the manufacturer's protocol. RT-PCR fragments were obtained by the use of forward primer PORCN-ex4F (5′-CACATGGTAGACACCGTAGACATGG-3′) and reverse primer PORCN-ex5R (5′-GGTAGCTGTGGAAGGATATCCAGG-3′) according to standard PCR protocols. PCR products of patient FDH5 were cloned into pCR2.1 TOPO TA Cloning Vector (Invitrogen). E.coli clones were subjected to colony PCR and PCR products from individual clones were sequenced.
X chromosome inactivation assay
Quantitative examination of the methylation pattern at the AR locus was performed as described earlier.10
Results
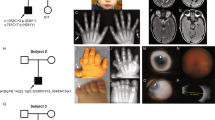
We performed mutation analysis of PORCN in 13 female patients with the clinical diagnosis of FDH (Figure 1) and four patients with MLS syndrome, previously tested negative for a mutation in HCCS. In total, we identified 12 different heterozygous intragenic mutations of PORCN in the 13 patients with FDH and no sequence variation in patients with MLS. Of these sequence alterations seven are nonsense and two missense mutations, two are small rearrangements, and one is a putative splice mutation (c.374-46T>A) (Table 2). Three of the mutations (p.W74X, p.R90X, and p.R243X) have been established as pathogenic sequence alterations,11, 12 whereas nine are novel (Table 2). In three individuals (FDH1, 8, and 10) with a novel mutation, we could demonstrate de novo occurrence of the mutation (including confirmation of paternity) (Table 2). For the remaining six individuals who carry none of the reported PORCN alterations, parental DNA was not available. Nonetheless, these mutations were not found in at least 204 control X chromosomes (Table 2).

Clinical features of FDH. Patients FDH11 (a–e), FDH1 (g), FDH6 (h–j), and FDH10 (k) show the typical clinical findings of FDH, such as dermal hypoplasia (leg of FDH 7 as newborn in (h) and at age 8 years in (i)), hyperpigmentation along the lines of Blaschko (c and i) and severe skeletal malformations, in particular syndactyly (d and e). Dysmorphic ears (b, j, and k) as well as dental defects such as oligodontia, hypodontia, enamel hypoplasia, and notched incisors (g) in FDH1. The father of patient FDH11 shows mild linear skin lesions on his calf (f).
The majority of patients were sporadic cases. However, in an affected mother–daughter duo (FDH3a and b) we detected the 2-bp duplication c.166_167dupAC in exon 3 and in patient FDH9, who was reported to have an affected mother and possibly also a maternal grandmother, the c.509G>A transition was identified which leads to a premature stop codon (p.W170X) (Table 2). Patient FDH11 was found to carry the c.1154G>A transition in the heterozygous state, predicting a substitution of the evolutionary highly conserved amino acid cysteine at position 385 by tyrosine (p.C385Y). The father of this patient had mild linear skin lesions on his calf (Figure 1f) and osteopathia striata, both clinical features typical of FDH suggesting somatic mosaicism. However, we could not detect the mutant base (adenine) in DNA extracted from blood cells of the father (no other material was available for testing).
In two individuals, FDH4 and 7 with c.268C>T (p.R90X) and c.1186C>T (p.R396X), respectively, the sequence profiles showed very low signals for the mutant variant superimposed on the wild-type sequence suggesting that the mutations are present in a mosaic state (Figure 2). The respective PCR product of each patient was subcloned and individual clones (PCR products) were subjected to SSCP analysis. We observed two banding patterns for the PCR products of each cloned amplicon. By DNA sequence analysis of selected clones we could assign the wild-type and the mutant sequence to either of the two banding patterns and identified a ratio of 9/53 (∼17%; mutant versus wild-type sequence) for the c.268C>T transition (FDH4) and 6/56 (∼11%) for the c.1186C>T alteration (FDH7) (data not shown), confirming the mosaic status of these patients.

Sequencing profiles from patients FDH4 (exon 3; left) and FDH7 (exon 14; right) with mosaic PORCN mutations. Top: profiles of directly sequenced PCR products suggestive of mosaicism for the c.268C>T mutation in FDH4 and c.1186C>T in FDH7. Middle: representative sequence of cloned PCR amplicons with wild-type sequence. Bottom: representative sequence of mutant cloned PCR products. The mutated and normal base is indicated by an orange arrow. Coding triplets and corresponding amino acid residues (one-letter code) are indicated.
In patient FDH5, we detected the only non-coding change, c.374-46T>A, in intron 4 which could create a novel splice acceptor site (TG>AG). FDH5 showed typical clinical features of FDH, such as linear skin lesions, microphthalmia, coloboma, syn- and polydactyly, nail dysplasia, cleft palate, auricle dysplasia, and microcephaly (Table 1). Therefore, we investigated the effect of this sequence change on splicing. In silico analysis using three splice site prediction programs revealed a low efficiency (if at all) for the normal acceptor site, whereas the newly introduced splice site was efficiently recognized by all programs (Figure 3a) suggesting that this site might also be used in vivo. Next, we analyzed PORCN transcripts by generating an RT-PCR product with primers located in exons 4 and 5 and obtained an amplicon with the expected size of 193 bp in patient FDH5 and two healthy individuals (Figure 3b). In addition, a larger PCR product of about 50 bp was observed in the patient and not in control individuals (Figure 3b). Subcloning and direct sequencing of both amplicons of FDH5 demonstrated splicing of exon 4 to exon 5 in the 193-bp amplicon (Figure 3c). However, an insertion of 44 bp between the last nucleotide of exon 4 and the first base of exon 5 was found in the larger PCR product (Figure 3d). These data show that use of the novel splice site located 46 bp upstream of the normal acceptor site in intron 4 causes an out-of-frame insertion of 44 intronic nucleotides in the PORCN mRNA.

The c.374-46T>A mutation in patient FDH5 introduced a novel splice acceptor site in PORCN intron 4. (a) Two of three splice site prediction programs failed to recognize the wild-type splice acceptor site, whereas the novel splice site yielded high scores by all three programs. (b) RT-PCR products were amplified from cDNA of patient FDH5 and two healthy individuals (C1 and C2) with primers located in PORCN exons 4 and 5. The normal 193-bp product is present in FDH5 and two controls, however, an additional amplicon of ∼240 bp was only amplified from the cDNA of FDH5 (white arrow). (c) Part of the sequence profile of the wild-type cloned RT-PCR amplicon (193 bp) of patient FDH5 showed splicing of exon 4 (ex 4) to exon 5 (ex 5). (d) Sequence analysis of the mutant cloned RT-PCR product (237 bp) revealed 44 bp of intron 4 spliced between exon 4 and 5. Coding triplets and corresponding amino acid residues (one-letter code) are indicated.
We analyzed the XCI pattern in the DNA isolated from leukocytes of all female patients with a sequence alteration in PORCN and identified random or slightly skewed (approximately 85:15) XCI (Table 2).
Discussion
We identified pathogenic mutations in PORCN in 13 female patients with FDH, with exons 3, 9, 10, and 12 representing mutational hot spots (this work and Grzeschik et al11, Wang et al12, Clements et al13, Leoyklang et al14). These data confirm that PORCN is the major gene, if not the unique one, for the classic FDH phenotype. Eleven of these patients appear to carry a constitutional mutation in the heterozygous state, whereas in two patients PORCN alterations represent a postzygotic mutation resulting in somatic mosaicism. De novo mosaic mutations have been suggested to be associated with a milder phenotypic outcome.11 Although this applies to patient FDH7 (Table 1), FDH4 with the mosaic nonsense mutation c.268C>T (p.R90X) showed the majority of FDH-typical characteristics (Table 1). These data suggest that a mosaic PORCN mutation found in leukocytes does not necessarily correlate with a mild phenotype in affected patients. Rather the relative abundance of the mutation in specific developing tissues, such as skin, bone, and eye seems to be relevant for development of a full-blown or mild phenotype. In the father of patient FDH11 with mild clinical features of FDH we could not detect somatic mosaicism of the missense mutation (c.1154G>A/p.C385Y) identified in his severely affected daughter. In contrast, in the affected father–daughter duo reported by Wang et al,12 the c.1059_1071dup13 mutation was found in the female patient with FDH in the heterozygous and in her mildly affected father in a mosaic state in DNA isolated from peripheral blood leukocytes. We propose that DNA isolated from other tissues than blood lymphocytes, if available, should be analyzed for the presence of a mosaic PORCN mutation in mildly affected cases with several typical features of FDH.
By PORCN transcript analysis we demonstrated in vivo recognition of a novel splice acceptor site 46 bp upstream of the natural acceptor site in intron 4 of patient FDH5 due to the c.374-46T>A mutation. Insertion of 44 intronic nucleotides in the PORCN mRNA most likely results in a functionally impaired protein harbouring 14 PORCN-unrelated amino acids after arginine 124 and lacking 338 PORCN residues at the COOH terminus. Although we did not determine the ratio (percentage) of the newly created splice site usage, this splice acceptor might be used with high efficiency based on the severe phenotype of FDH5 (Table 1) and its efficient recognition by three splice prediction programs (Figure 3a).
We identified random or slightly skewed XCI in our patients with a heterozygous intragenic PORCN mutation that is in line with random XCI in the majority of mutation-positive patients with FDH.11, 12, 14 This data suggests that PORCN does not seem to be critical for cell division or survival, otherwise cells with the mutated X chromosome active would fail to divide or survive, eventually leading to severe skewing of XCI. However, in female patients with a microdeletion covering PORCN skewed XCI has been consistently observed.11, 12, 20 Nonetheless, this finding is not surprising as one or more of the neighbouring genes (proteins) in the deleted interval, such as SLC38A5, FTSJ1, or OATL1 could account for selective survival of cells with the wild-type X chromosome active.11, 12 Indeed, skewed XCI has been found in a healthy female patient who carries a heterozygous 50-kb deletion encompassing the two genes SLCA38A5 and FTSJ1.21 This data suggests that lack of SLCA38A5 or FTSJ1 cause a growth disadvantage of cells with the mutation on the active X chromosome leading to skewed XCI in female patients heterozygous for a mutation in SLCA38A5 or FTSJ1. It has been proposed that the pattern of XCI, not only in blood cells but also in other tissues influences the development of clinical features in female patients with an X-linked male lethal disorder resulting in a high phenotypic variability (from the most severe phenotype to a milder manifestation or the total absence of clinical features).19 Similarly, the phenotype also varies in female patients with a microdeletion including PORCN (as well as SLCA38A5 and FTSJ1) and skewed XCI. Typical clinical features of FDH, such as linear skin lesions, dermal hypoplasia, nail dystrophy, and asymmetric bone defects, but no ocular abnormalities have been observed in five sporadic female patients with a submicroscopic deletion encompassing PORCN and additional genes.11, 12 A 112-kb deletion including PORCN was found in four female patients from a three-generation family with localized subepidermal telangiectasias following Blaschko's lines (angioma serpiginosum), a rare skin disorder that has been suggested to be allelic to FDH.20, 22 Taken together, female patients with an intragenic PORCN mutation and random XCI are supposed to show the classical FDH phenotype. In these cases, somatic mosaicism can lead to a milder phenotype. In contrast, female patients with a microdeletion covering PORCN and another gene, that causes selective growth disadvantage when absent, may show higher phenotypic variability, including unusual mild phenotypes that could be due to severe skewing of XCI. Thus, the phenotype in female patients heterozygous for a pathogenic X-linked mutation can be influenced by several different mechanisms, such as factors related to the X inactivation process,23, 24, 25 somatic mosaicism,11, 12 and the nature of the mutation,24 suggesting that establishing a genotype–phenotype correlation is difficult, if not impossible, in these disorders.
One important finding of this study is the absence of a PORCN mutation in patients with MLS syndrome, who had been tested negative for mutations in the established disease gene HCCS. Two of these patients (MLS1 and 3) did not show the classical MLS phenotype as microphthalmia was absent. However, the presence of other clinical features, such as corpus callosum hypo- or aplasia, cardiomyopathy, and diaphragmatic hernia strongly suggested that the phenotype of these patients belongs to the MLS spectrum. Although we cannot exclude the presence of cryptic alterations such as changes in an intron, the 5′- or 3′-untranslated region, or defects in the promoter region or cis-regulatory element of either HCCS or PORCN, our negative findings indicate that mutations of PORCN are not involved in the aetiology of MLS. Thereby, we confirm the concept that MLS and FDH are not allelic and also exclude the possibility that one of the four patients had FDH rather than MLS. In addition, severely skewed XCI in the four PORCN- and HCCS-mutation negative individuals with a phenotype strongly resembling MLS may provide at least some evidence for X-linked genetic heterogeneity in this disease.
Taken together, our data suggest that (i) FDH is a clinically and genetically homogeneous disorder and in patients with a clear-cut FDH phenotype a PORCN mutation most likely will be identified, (ii) somatic mosaicism can be present in patients with a mild or classic FDH phenotype and particular attention should be paid to this phenomenon when diagnostic testing is performed, (iii) PORCN deficiency itself does not cause lethality of leukocytes with the mutation on the active X chromosome, and (iv) MLS and FDH are different entities, both from the clinical and genetic point of view.
Accession codes
References
Goltz RW, Peterson WC, Gorlin RJ, Ravits HG : Focal dermal hypoplasia. Arch Dermatol 1962; 86: 708–717.
Goltz RW : Focal dermal hypoplasia syndrome. An update. Arch Dermatol 1992; 128: 1108–1111.
Van den Veyver IB : Microphthalmia with linear skin defects (MLS), Aicardi, and Goltz syndromes: are they related X-linked dominant male-lethal disorders? Cytogenet Genome Res 2002; 99: 289–296.
Ballabio A, Andria G : Deletions and translocations involving the distal short arm of the human X chromosome: review and hypotheses. Hum Mol Genet 1992; 1: 221–227.
Lindsay EA, Grillo A, Ferrero GB et al: Microphthalmia with linear skin defects (MLS) syndrome: clinical, cytogenetic, and molecular characterization. Am J Med Genet 1994; 49: 229–234.
Naritomi K, Izumikawa Y, Nagataki S et al: Combined Goltz and Aicardi syndromes in a terminal Xp deletion: are they a contiguous gene syndrome? Am J Med Genet 1992; 43: 839–843.
Donnenfeld AE, Graham Jr JM, Packer RJ, Aquino R, Berg SZ, Emanuel BS : Microphthalmia and chorioretinal lesions in a girl with an Xp.22.2-pter deletion and partial 3p trisomy: clinical observations relevant to Aicardi syndrome gene localization. Am J Med Genet 1990; 37: 182–186.
Happle R, Daniels O, Koopman RJ : MIDAS syndrome (microphthalmia, dermal aplasia, and sclerocornea): an X-linked phenotype distinct from Goltz syndrome. Am J Med Genet 1993; 47: 710–713.
Mücke J, Happle R, Theile H : MIDAS syndrome respectively MLS syndrome: a separate entity rather than a particular lyonization pattern of the gene causing Goltz syndrome. Am J Med Genet 1995; 57: 117–118.
Wimplinger I, Morleo M, Rosenberger G et al: Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome. Am J Hum Genet 2006; 79: 878–889.
Grzeschik KH, Bornholdt D, Oeffner F et al: Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet 2007; 39: 833–835.
Wang X, Reid Sutton V, Omar Peraza-Llanes J et al: Mutations in X-linked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat Genet 2007; 39: 836–838.
Clements SE, Wessagowit V, Lai-Cheong JE, Arita K, McGrath JA : Focal dermal hypoplasia resulting from a new nonsense mutation, p.E300X, in the PORCN gene. J Dermatol Sci 2008; 49: 39–42.
Leoyklang P, Suphapeetiporn K, Wananukul S, Shotelersuk V : Three novel mutations in the PORCN gene underlying focal dermal hypoplasia. Clin Genet 2008; 73: 373–379.
Takada R, Satomi Y, Kurata T et al: Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev Cell 2006; 11: 791–801.
Willert K, Brown JD, Danenberg E et al: Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003; 423: 448–452.
Morleo M, Pramparo T, Perone L et al: Microphthalmia with linear skin defects (MLS) syndrome: clinical, cytogenetic, and molecular characterization of 11 cases. Am J Med Genet A 2005; 137: 190–198.
Kherbaoui-Redouani L, Eschard C, Bednarek N, Morville P : (Cutaneous aplasia, non compaction of the left ventricle and severe cardiac arrhythmia: a new case of MLS syndrome (microphtalmia with linear skin defects)). Arch Pediatr 2003; 10: 224–226.
Morleo M, Franco B : Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J Med Genet 2008; 45: 401–408.
Houge G, Oeffner F, Grzeschik KH : An Xp11.23 deletion containing PORCN may also cause angioma serpiginosum, a cosmetic skin disease associated with extreme skewing of X-inactivation. Eur J Hum Genet 2008; 16: 1027–1028.
Froyen G, Bauters M, Boyle J et al: Loss of SLC38A5 and FTSJ1 at Xp11.23 in three brothers with non-syndromic mental retardation due to a microdeletion in an unstable genomic region. Hum Genet 2007; 121: 539–547.
Blinkenberg EO, Brendehaug A, Sandvik AK, Vatne O, Hennekam RC, Houge G : Angioma serpiginosum with oesophageal papillomatosis is an X-linked dominant condition that maps to Xp11.3-Xq12. Eur J Hum Genet 2007; 15: 543–547.
Franco B, Ballabio A : X-inactivation and human disease: X-linked dominant male lethal disorders. Curr Opin Genet Dev 2006; 16: 254–259.
Migeon BR : Why females are mosaics, X-chromosome inactivation, and sex differences in disease. Gend Med 2007; 4: 97–105.
Van den Veyver IB : Skewed X inactivation in X-linked disorders. Semin Reprod Med 2001; 19: 183–191.
Acknowledgements
We are grateful to the patients and their parents. The results summarized here are part of the MD thesis of May-Britt Harmsen at the University of Hamburg. We thank Inka Jantke for skillful technical assistance, Isabella Wimplinger for sequence analysis of HCCS and X chromosome inactivation analysis, Manuela Morleo and Brunella Franco for copy number analysis of HCCS exons, and Stephan Schneider for his constant support to obtain material from patient FDH5.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Harmsen, MB., Azzarello-Burri, S., García González, M. et al. Goltz–Gorlin (focal dermal hypoplasia) and the microphthalmia with linear skin defects (MLS) syndrome: no evidence of genetic overlap. Eur J Hum Genet 17, 1207–1215 (2009). https://doi.org/10.1038/ejhg.2009.40
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.40
Keywords
This article is cited by
-
Goltz-Gorlin Syndrome: Revisiting the Clinical Spectrum
The Indian Journal of Pediatrics (2018)
-
Expanding the phenotypic spectrum of PORCN variants in two males with syndromic microphthalmia
European Journal of Human Genetics (2015)
-
Novel Mutation in a Child with Goltz Syndrome
The Indian Journal of Pediatrics (2012)
