
Abstract
Age-related macular degeneration (AMD) is a late onset vision disorder. Recent studies demonstrate that alterations in complement cascade genes are associated with AMD. Of the three identified complement loci, variants in complement factor H (CFH) have the highest impact as does an independent locus at 10q26. Our matched case–control study using the Age-Related Eye Disease Study (AREDS) cohort confirms and extends the associations in these loci. Subjects were genotyped for single nucleotide polymorphisms (SNPs) from CFH, complement component 2 (C2), complement component 3 (C3), complement factor B (CFB), age-related maculopathy susceptibility (ARMS2), HtrA serine peptidase 1 (HTRA1), and apolipoprotein E (APOE). Individual SNPs, and haplotypes showed risk trends consistent with those seen in other population studies for CFH, C3, C2, and CFB. SNP rs10490924 on chromosome 10 in exon 1 of the ARMS2 gene showed a highly significant association with an odds ratio (OR) of 3.2 (95% CI 2.4–4.2) for the risk allele and rs11200638 located in the proximal promoter region of HTRA1 showed a higher significant association with an OR of 3.4 (95% CI 2.5–4.6) with our AMD cases. We found that APOE haplotypes were not significantly associated with disease status. Adjustments for other risk factors did not significantly alter the observed associations. This study validates the complement pathway's involvement in AMD and suggests that allelic variants in complement genes have a direct role in disease. These results also support previous findings that variants in the region of 10q26 exert an independent risk for AMD.
Similar content being viewed by others

Introduction
Age-related macular degeneration (AMD) is a late onset retinal disease, which is the most common cause of severe vision loss of the elderly in the US, and there are indications that prevalence rates are rising. These rates combined with an aging US population have prompted tremendous research efforts into finding both the environmental and genetic contributions to AMD, many of which have yielded substantial knowledge and understanding of the etiology of the disease.
Familial studies show that among first-degree relatives of probands there is a three-fold risk for developing AMD.1, 2 However, as this disease has a late onset, family-based studies are difficult to conduct, making case–control studies the design of choice for most researchers. Linkage and whole genome association studies have narrowed the genetic focus to a few chromosomal regions that are highly associated with AMD.3, 4, 5, 6, 7, 8 Inflammatory regulation of the complement pathway has been established as having a central role, and genetic variants coding for the proteins within these pathways are associated with increased and decreased risk.7, 9, 10, 11 Two variants located at 10q26 have been shown to be very strongly associated with AMD in virtually all studies to date.3, 12 The genes encoded in this region have not yet been fully characterized and their precise function is still unknown.
This study was aimed at investigating single nucleotide polymorphisms (SNPs) and common haplotypes in complement factor H (CFH), including a deletion located downstream of CFH,6, 13 complement factor B (CFB), complement component 2 (C2), and complement component 3 (C3) involved in the complement cascade, and variants in age-related maculopathy susceptibility (ARMS2) and HtrA serine peptidase 1 (HTRA1) using a carefully matched subset of the Age-Related Eye Disease Study (AREDS) case and control subjects. In addition, using analysis techniques beyond logistic regression, we present a means for assessing epigenetic effects between unlinked loci using a genetic algorithm and regression tree analysis, tools that assess the contributory effects of each individual genotype in an additive model. Consequently, we account for ∼80% of the cases and ∼50% of the controls on the basis of these models.
The confirmation that these genes are associated with AMD will contribute to the growing breadth of knowledge on its pathogenesis; and this insight can be used to improve early detection in high-risk individuals and provide a logical basis for future clinical trials and preventive screening in high-risk cohorts.
Methods
Study population in the AREDS cohort
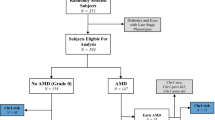
Individuals selected for this study were participants in the AREDS, which was initially conceived as a long-term multi-center, prospective study of the clinical course of AMD and age-related cataract. AREDS also included a clinical trial of high-dose vitamin and mineral supplements for AMD and a clinical trial of high-dose vitamin supplements for cataract.14 Briefly, patients over the age of 55 years who self-reported to participating eye clinics between 1992 and 1996 were considered for AREDS, and eligibility was based on AMD symptoms and retinal photographs. A total of 2097 patients were selected and enrolled in the clinical trial, all were put into one of five AMD severity categories, and then randomized into intervention and non-intervention groups. In this sample set, 215 patients who were free of AMD symptoms acted as controls, followed for observation only. The full details and results of the original AREDS study are reported elsewhere.15, 16
Observational study
For our case–control observational study, we obtained validated DNA samples of all the available AREDS participants from the National Eye Institute-AREDS Genetic Repository at Coriell Institute for Medical Research (Camden, NJ, USA). We matched two cases with one control on sex and race; and the difference in mean age between cases and controls was <2 years. This design was most suitable because of the small number of controls available to us because AREDS was designed as a prospective cohort study with an intervention, and only 215 disease-free patients were included as a control group. Because of this limit, a 2:1 match was favored over a 1:1 match for the simple fact that it gave us more statistical power, bringing our total sample to 639 patients.
Selection criteria for our cases and controls
We relied on the five AMD categories created and utilized in the original AREDS clinical trial to define our cases and controls.17 A brief description of these categories is as follows: category one was described as being completely free of AMD, and all our controls were selected from this group. Category two subjects were ambiguous, meaning that they did not have any clear symptoms of early-stage AMD, but also could not be considered free from eye disease symptoms, and so they were identified as ‘other’; these individuals did not qualify as an AMD case or a control by our definition and were excluded from our study. Category three subjects, having early-stage AMD, had many medium-sized drusen or one or more large drusen in one or both eyes. Category four patients had end-stage AMD in one eye; and subjects in category five had end-stage AMD in both eyes. All of our cases were selected from categories three, four, and five.
Genotyping
SNPs were genotyped using Applied Biosystems’ Taqman® 5′ nucleotidase assays, Taqman Universal PCR Master Mix: no AmpErase UNG, and 5 ng of genomic DNA. The thermal cycling conditions consisted of an initial hold at 95°C for 10 min followed by 50 cycles of a 15 s 95°C denaturation step and a 1-min 60°C annealing and extension step. A 7900HT instrument was used to detect fluorescent probes, and Sequence Detection Software (SDS) v2.2 was used to discriminate alleles and call genotypes (Applied Biosystems, Foster City, CA, USA).
The deletion distal to CFH at 1q31.3 was detected using a robust, duplex PCR/gel assay, which was designed and developed by us (see Figure 1). The PCR protocol consists of two pairs of primers, the first pair annealing within the imputed deletion producing a band of 529 bp on a 2% agarose gel, and the other pair simultaneously amplifying a region at 11q22.3, which consistently produces a 359 bp amplicon regardless of deletion status, serving as a PCR quality control. This banding pattern, one band for homozygous deleted subjects and two bands for non-deleted subjects, permitted visual scoring and compilation of results into a 2 × 2 contingency table.

Agarose gel image showing a single band for deletion carriers and double bands for non-deleted individuals. As described in Methods, this duplex consists of two pairs of primers in a single PCR reaction. The first pair anneals at a region at 11q22.3 and acts as the control, consistently producing the 359-bp lower band in all subjects regardless of deletion status. The second pair anneals within the deletion and will only amplify if that region is not deleted, producing the 529-bp upper band. Subjects who have the deletion display a single band, and non-deleted subjects display two bands. PCR products were subjected to electrophoresis on a 2% TAE gel, run at 100 mV for 1 h, and stained with ethidium bromide.
Microarray concordance tests
We made efforts to reconcile the 111 950 SNPs we mapped to the Affymetrix Microarray and the 108 844 SNPs we mapped to the Illumina BeadArray with the 16 SNPs we genotyped using the 5′ nucleotidase assay. We found no overlap between the SNPs typed using the Illumina BeadArray and the Affymetrix microarray on the AREDS samples. However, we were able to cross-check two SNPs, rs2230199 and rs800292, typed on the Illumina BeadArray with our TaqMan 5′ nucleotidase genotypes, and three SNPs: rs2736911, rs10490924 and rs10490923 to the Affymetrix Microarray. Of the 536 genotype calls susceptible to cross-check in the Affymetrix data, there were four discordant calls and two 5′ nucleotidase no calls, for a total concordance rate of 98.8%. With respect to the Illumina data, there were 410 genotype calls susceptible to cross-check, with four discordant calls and nine no calls, of which five were in the Illumina data, for a final Illumina-TaqMan concordance rate of 96.8%. This level of concordance in unselected samples that were cross-checked only after our 5′ nucleotidase typing was complete and analyzed supports the conclusions drawn in this paper.
Statistical analysis
We matched AREDS cases and controls as closely as possible on baseline characteristics, including age, sex, and race, and differences between groups were tested using the χ2 test for categorical variables (see Table 1). For all polymorphisms, we did an analysis of allele frequencies in a 2 × 2 contingency table using the χ2 test or the two-tailed Fisher's exact test (if <5 patients in one cell), looking for an association between the risk allele and AMD case status. These data were then analyzed by genotype using unconditional logistic regression with case status (0/1) as the outcome, genotypes (11, 12 and 22) as categorical variables, and age as a continuous variable; and we report odds ratios (ORs), 95% confidence intervals, and two-sided P-values. For the reference group, we decided to consistently use the ancestral allele as reported by NCBI in all our loci, even though in some it was not the most frequent allele in Caucasians. Estimated haplotypes were analyzed using logistic regression as categorical variables. To assess for potential confounders, we used clinical and questionnaire data provided to us by the NEI, including tobacco use, aspirin use, vitamin supplement use, marital status, highest level of education completed, BMI, blood pressure, and history of cancer. To assess possible interaction and/or effect modification between Y402H and our other SNPs, a separate logistic regression analysis was performed for each polymorphism and adjusted for Y402H alleles. Genotype distributions for each polymorphism were tested for deviation from Hardy–Weinberg equilibrium using χ2. Because of the low frequency of the mutant allele in the four SNPs in C2 and CFB (rs9332739, rs547154, rs4151667, and rs4151659), we pooled heterozygous and homozygous mutant genotypes (12 and 22) and compared this group to the reference genotype.
For haplotype estimation, we used PHASE version 2.11 (Matthew Stephens, University of Washington, Seattle); STATA 9 (StataCorp LP, College Station, TX) was used for the statistical analysis.
Multilocus contribution estimation methods
A matrix format, allele-based genotype data set for Sapio Exemplar 4.0.8 (York, PA) was created from the complete genotypes (Excel spreadsheet available at ftp://ftp.ncifcrf.gov/pub/users/goldb/AMD2009). In total, 3000–5000 models with node sizes from 3 to 5 at 20 iterations each were created using the genetic algorithm module. A G-test, Fisher's exact test, or χ2 test was used to test the genetic algorithm model fit with the learning set. Two-thirds of the cases and controls, randomly presented, were used for the learning set. The test set was the remaining one-third of cases and controls. Statistics are presented for both the learning set and the test set in results and discussion. Separately, the same data set was provided as input for SPSS version 14.0 (Chicago, IL) complete with the Classification Tree 13.0 module. SPSS was instructed to provide an exhaustive chi-squared automated interaction detector (CHAID) with case–control status as the dependent variable with a maximum tree depth of three, a minimum of 100 cases in the parental node, and no fewer than 50 cases in the child nodes. Finally, we tested the strength of the models by randomly swapping case–control identifiers to make 1000 input sets of genotype data equivalent to our actual set. These were processed into models and assessed through statistical testing in both Sapio Exemplar and SPSS version 14.0.
Results
Participants
This sample of AREDS is predominantly Caucasian, approximately 50% female, and roughly half of both cases and controls are current or past smokers. Mean age of cases matched the mean age of controls by less than 2 years (Table 1). The controls and the cases were within Hardy–Weinberg limits for all polymorphisms.
Association of CFH variants and the CFHR1/3 deletion
To determine the association of CFH region variants, the genotype and allele frequencies for three polymorphisms in the CFH gene (rs800292, rs1061170 and rs1410996) were determined. These variants were all significantly associated with AMD case status under a multiplicative genetic model (r fold increased risk for one risk allele and r2 increase in the risk for two risk alleles) as shown in Tables 2 and 3. The deletion located approximately 6400 bp 3′ to CFH, which removes CFHR3 and CFHR1 entirely,6, 13 was analyzed using a 2 × 2 contingency table and Fisher's exact test (Figure 1). This deletion is highly protective6, 9 and was also significantly associated with a decreased risk in AREDS (OR=0.19; Table 4). Haplotype frequency estimates for the CFH polymorphisms and the deletion are shown in Table 5. One very high-risk haplotype (GCGN) was found in 58% of cases versus 35% in controls and is associated with a three-fold increased in risk compared with the haplotype 1 reference (OR=3.37). Copy number for this haplotype also showed a graded increase in risk, one copy elevating the OR to 2.2 and two copies elevating the OR to 6.6, and also when compared with all other haplotypes (data not shown). Another common haplotype contains the CFHR1 → CFHR3 deletion (GTAD) and is highly protective with an OR of 0.34 as compared with the reference. This protective haplotype was analyzed further in a 2 × 2 table and compared with all other haplotypes and was also shown to be highly protective (data not shown).
Variants in CFB and C2
To determine the contribution of variants in the C2 and CFB genes, five SNPs were typed in the AREDS cohort (Tables 2 and 3). Four out of the five SNPs we typed in this chromosomal region are rare variants, and the genotype analysis was performed by pooling the heterozygote and homozygote mutants together and assuming a dominant genetic model; all but one is significant (Table 2). Because of their close proximity to each other in the genome, we estimated haplotype frequencies using the five SNPs from the two genes. As shown in Table 5, haplotype 6 (CAAAC) is associated with a decrease in risk, and to a lesser degree, haplotype 4.
C3: a key protein in complement regulation
SNPs in C3 have been found to be significant in some studies of AMD.18 Of the two SNPs we typed in C3, rs2230199 was moderately associated with AMD cases with an OR of 1.9 for heterozygous (CG) and 2.5 for homozygous mutant (GG) (Table 2). Likewise, a similar magnitude of association was observed when we combined homozygous mutant genotypes with heterozygous genotypes (CG+GG), and also when analyzed by alleles (OR=1.8, 95% CI 1.4–2.5). The promoter region SNP, rs171094, was slightly significant when analyzed by genotype and allele. Haplotype frequencies for C3 are shown in Table 5. The two risk haplotypes, though low in overall counts, both contain the high-risk allele for rs2230199.
Analysis of the 10q26 ARMS2 and HTRA1 locus
The chromosome 10q26 locus containing the ARMS2 and HTRA1 genes is highly associated with AMD, although the causative gene and variants are unproven.19 We confirmed previous reports of the very strong association on chromosome 10 by typing rs10490924 in ARMS2, and rs11200638 in the promoter region of HTRA1. These SNPS are in high linkage disequilibrium.20 Both showed a similar magnitude in risk associated with disease status when analyzed by genotype and by allele count. For rs10490924, carriers of one risk allele (GT) had an elevation in the OR to 3.1 (95% CI 2.1–4.5), whereas carriers of two risk alleles (TT) had an OR of 9.2 (CI=3.9–21). Analyzing this SNP by alleles was more robust and gave it more power, resulting in an OR of 2.7 and a tighter confidence interval (CI=1.9–3.8) for the T risk allele. See Tables 2 and 3 for genotypes and alleles for all SNPs. The promoter SNP in HTRA1, rs11200638, being in high LD with rs10490924, was also strongly associated with AMD and showed a similar pattern of risk: genotype analysis showed that carriers with one risk allele (GA) had an elevated OR of 3.2 (CI=2.1–4.6) and those carrying two risk alleles (AA) had an OR of 9.1 (CI=4.1–20). Again, a more robust and powerful analysis method of allele count was appropriate for this SNP given the small number of AA homozygotes, which yielded an OR of 3.4 (CI=2.5–4.5). Of note, the other two SNPs typed in ARMS2, rs10490923 and rs2736911, showed protective trends when analyzed individually by genotype and by alleles, but were not statistically significant.
Table 5 contains the results of haplotype analysis for chromosome 10 polymorphisms. We estimated four common haplotypes from these variants in ARMS2 and HTRA1, and the unfavorable haplotype 4 was seen in 39% of cases and only 16% of controls, and showed an OR of 3.4 (CI=2.5–4.7). Clearly the risk in this haplotype is attributed to the alleles of rs10390924 and rs11200638.
No association with apolipoprotein E
Some previous studies have found a correlation with the apolipoprotein E (APOE) haplotype and AMD disease.21 Using logistic regression, we analyzed APOE haplotypes as categorical predictors to generate ORs. We did not see a statistically significant result for these variants in our sample of AREDS. We performed a separate analysis in which all our other loci were regressed and adjusted for APOE haplotypes. Very slight differences were seen between unadjusted and adjusted OR among all our polymorphisms, and none changed significance level to any degree (data not shown).
The effect of smoking on AMD
Several epidemiology studies have found an association between smoking and increased risk of AMD.21 A separate analysis was carried out for all our loci using logistic regression while adjusting for smoking. Very weak interactions were seen, but smoking did not appear to change risk profiles remarkably and did not change statistical significance levels for these specific SNPs (data not shown).
Results of multistate modeling
To estimate the fraction of cases and controls that can be explained by the data, multistate models of the genotypes constructed using a genetic algorithm and, separately, a CHAID model. The mechanism of model construction markedly differs between these two methods: the first requires a ‘training set’ and then tests the outcomes on a ‘test set’, and the second provides only a χ2 test-based principal component stratification. The models agreed that the major contributions to the phenotype are from the CFH region on chromosome 1 and the HTRA1-ARMS2 region on chromosome 10. The GA model concluded that a protective genotype at the CFB-C2 locus11 is of sufficient consequence to include in the interaction model, whereas the CHAID method did not find this. Neither modeling method found C3, the CFHR1-CFHR3 deletion alone, or the APOE genotype to account for a significant number of cases and controls (Figure 2). Permutation testing of the models using 1000 simulated data sets where the case or control identifiers had been randomly swapped resulted in χ2 value distributions that did not overlap those observed on the verified data set.

Multistate model of AMD data. A representation of multistate modeling using the (a) genetic algorithm method as implemented in Sapio Sciences Exemplar Genetic Analysis platform and (b) a chi-squared automated interaction detector (CHAID) model as implemented in SPSS Classification Trees module in version 14.0 is shown. In (a), data were provided on 624 research subjects at 17 loci in addition to the CFHR1-CFHR3 deletion and the APOE haplotype. The genetic algorithm was then run on a ‘training set’ of 413 research subjects and then subsequently used to predict affection status in the remaining 211 research subjects. The genetic algorithm model represented in (a) determined that if rs1061170 encoding Y402H in CFH on chromosome 1 is CC AND rs547154, an intronic SNP in the C2 gene on chromosome 6 is CC, then 160/421 cases are explained. Alternatively, the genetic algorithm model states that if an A allele is present at rs11200638, which is 5′ to HTRA1 on chromosome 10, then 320/421 cases are explained. In (b), the same data were presented to SPSS program, which found initial stratification by rs1410996 an intronic SNP in CFH on chromosome 1 with CC genotype determining case status in 256/421 cases. Among these cases, only 91 are devoid of the predisposing allele A at rs11200638, which again is 5′ to HTRA1 on chromosome 10. The status of 339 cases and 99 controls is accounted for using rs1410996 an intronic SNP in CFH on chromosome 1, rs11200638, which is 5′ to HTRA1, and rs10490924, which is A69S in ARMS2, the latter two in chromosome 10, using the CHAID model. ***P<10−3.
Discussion
In a matched sample set from the AREDS cohort, we observed associations between haplotypes in genes involved in the complement cascade, and also in two genes on chromosome 10, with AMD. Expectations about the risk or protective effects of each were consistently met in our analysis, which reliably validates and supports numerous other research studies of AMD. Hypotheses about the nature of the associations of these SNPs and AMD are discussed below.
The physiological role of CFH is to downregulate and control initiation and magnitude of the complement cascade, and its function is dependent on how well it binds to cell surfaces. CFH consists of 20 short consensus repeat (SCR) modules, and SCR7 and SCR20 specifically bind to polyanions on the surface of cells.22 It is the SCR7 that harbors the polymorphic residue Y402H, and the His402 allele alters the precise specificity and affinity of its interaction with the cell surface, leading to altered levels of CFH retention on the retinal/macular surface, and thereby affecting complement activity in the area.22 This effect is supported by our study of polymorphism rs1061170 (Y402H) in which we see an OR of 2.0 in patients with one copy and 6.2 in patients with two copies of the His402 variant. Other studies have suggested that this polymorphism accounts for up to 50% of the population attributable risk for AMD.7, 8, 9, 10, 12 The other missense mutation in CFH, rs800292, changes protein residue 62 from Ile to Val, but the resulting structural changes and effects are understudied. Both rs800292 and the non-coding rs1410996 showed unfavorable odds of disease in a recessive manner. That is, we did not see a statistically significant difference in these two for the heterozygous genotype, whereas we did for homozygous mutants (ORs=3.7 and 6.6, respectively). Haplotype analysis of this gene allowed us to assess the effect of all the coding variants together. The presence of a risk allele from rs1061170 in the high-risk haplotype (GCGN) clearly confers a significantly increased risk for AMD.
A large deletion 3′ to CFH is associated with protection from AMD. The CFHR1 and CFHR3 genes deleted in this region are closely related to CFH and their proteins are abundant in serum, but they have as yet no defined role in the complement cascade and no clear function. Rarely did the deletion segregate with the high-risk C allele for Y402H in our sample set, and so its protective effect could not be independently assessed against the risk exerted by a mutated CFH gene. Unfortunately, we were not able to distinguish the copy number for the deletion, which could change the haplotype estimations, and thus our risk assessments. Future plans are to develop a method of distinguishing heterozygous (+/−) from homozygous (+/+) non-deleted individuals.
CFB is an alternative complement cascade protein. Activation of the alternative pathway is initiated by cleavage of C3b-bound factor B (CFB), resulting in the formation of the C3Bb complex (C3 convertase). Of the two SNPs we studied in CFB, only rs4151667 showed a statistically significant difference between cases and controls. This SNP gives rise to a missense variant that results in amino acid 9 changing from Leu to His in the signal sequence region of the protein. There appears to be some interaction between one SNP in CFB and Y402H in CFH. The unadjusted OR for rs2072633 is not statically significant alone (P=0.63), but after adjusting for Y402H it becomes significant (P=0.01). This suggests CFB effectively cancels the aberrant activity of a mutated CFH gene, thus modifying risk in a favorable direction; however, it does not reduce risk further beyond a wild-type 402Y phenotype. Also of note, because C2 and CFB are closely related in structure and function, rs4151667 from CFB and rs9332739 from C2 show the same frequency of common and rare alleles between cases and controls, resulting in very consistent protective effect between them. In fact, as we reported previously, the C2 and CFB genes are located only 500 bp apart on chromosome 6p21 in the major histocompatibility complex class III region, and are evolutionarily and structurally closely related.11 Previous genetic studies have shown that mutations in C2, as in CFB, can compromise the efficiency of the complement cascade. The variants in C2 show a protective effect in our study (Table 2), but it is unclear whether the association is in any way causal. Studies of drusen composition have found deposits of proteins from the alternative complement pathway, but their analogs from the classical pathway, including C2, have not been found. Analysis of haplotypes across this region shows two that are protective, both of which appear to be due to the variants from CFB.
C3 is the most abundant complement component, synthesized predominantly in the liver but to a lesser extent in other cells and tissues. Significant C3 transcripts are detectable in the neural retina, choroid, retinal pigment epithelium, and cultured retinal pigment epithelium cells. In contrast to the other complement cascade proteins showing a protective effect, variants in C3 increase the risk for AMD. Experimental evidence of functional differences between wild-type versus mutated alleles in C3 is not conclusive; however, this gene is mutated in many other diseases affecting epithelial tissues that are very similar in structure to choriocapillaris, Bruch's membrane, and retinal pigment epithelium. It is unclear how a mutated form of this gene would lead to sequestered complement protein deposits in some kidney disorders, but it is strikingly similar to the drusen deposits seen in the dry form of AMD. Functional studies are needed to further characterize this important protein and how it contributes to the incremental risk associated with its variants.
Polymorphism at 10q26 spanning ARMS2 and the promoter region of HTRA1, as well as the estimated haplotypes from these variants, shows very strong association with AMD case status. Our sample showed a slightly higher risk from rs11200638 in HTRA1, yet much attention and focus remain on rs10490924 in ARMS2; Fritsche et al,23 extensively sequenced several individuals with the ARMS2-HTRA1 risk haplotype and found non-sense-mediated decay of ARMS2 resulting from a complex variant in the 3′ UTR (c.*372_815delins54) and proposed this as the etiologic variant. On the other hand, a non-sense allele in ARMS2 (R38X) does not appear to confer significant risk (suggesting that a loss of function is not responsible for the ARMS2-deficient etiology23 and data not shown). Thus, confirmation that the c.*372_815delins54 is the functional variant remains elusive.19
The APOE gene has been found to be associated with AMD in some but not all studies.24, 25 As can be seen in the data available at our FTP site, there were a total of 132 E4 allele-bearing subjects in the present study. A two-sided test for association yielded a P-value of 0.097. It could be argued that a one-sided P-value is more applicable; however, that value was also not significant at P=0.054. Yet, there were only seven homozygous E2 individuals and five homozygous E4 individuals in our study. An additional nine individuals were E2/E4. Thus, we conclude that we did not have power comparable to that of Schmidt et al,26 who examined 1260 research subjects to come to the conclusion of association.
Recently, Yang et al27 reported the TLR3 SNP, rs3775291, to be very protective against geographic atrophy but not other AMD categories. This gene, which encodes a viral sensor that supports innate immunity and host defense,27 is a plausible candidate for AMD etiology. Edwards et al28 found a borderline association with rs3775291, in some cohorts and negative results in others, and we and others have failed to replicate this association, nor did it contribute to our multistate modeling.29
Surprisingly, we did not see our risk profiles change appreciably when we adjusted for smoking status, where most other studies show smoking to be a risk factor.30 There were 46 never smoker individuals in our research study, of which 32 were cases and 14 were controls. Although we saw no statistically significant relationship, this may be on account of our age choice of samples: There were no never smoker controls below the age of 60 years with whom to compare the cases. Among the cases below the age of 60 years, 14 were past smokers, 18 were current smokers, and 7 were never smokers. Therefore, our sample size, ascertainment, and paucity of younger controls make it inappropriate to come to a conclusion concerning smoking and AMD. Other reasons that we did not see a higher risk in smokers are that we did not stratify by AMD disease categories as some researchers have done, nor did we adjust for smoking in the haplotype analysis. We were limited to three categories of smoker by the AREDS self-reported questionnaire.
Our multistate modeling, using two independent analytic approaches on the same data set, concluded that we could account for ∼80% of the cases and ∼50% of the controls on the basis of the CFH locus, the chromosome 10 gene associated SNPs, and one C2 intronic SNP on chromosome 6. We realize, however, that the size of this cohort and number of controls may be insufficient to make a definitive interaction assessment. To this caution, we add the confounding effects of population stratification and a small but consequential effect of missing genotypes. In addition, new statistical methods for assessing interactions are emerging; thus, our current assessment is no more than a best guess estimate. On the other hand, we believe it sufficiently meritorious and diligent to report in this paper at this time.
Identifying genetic risk factors for AMD is important for understanding the etiology of the disease. Currently, AMD is not preventable and its causes are not fully understood although a number of risk factors and a few protective factors have been identified. A better understanding of the pathology and genetic predisposition of AMD is vital for both the affected individual and the larger public's health. In the coming years, we will have a greatly increased population over the age of 65 years with greater longevity, and therefore AMD disease will continue rising as an urgent public health issue. Vision loss resulting from AMD will have a considerable impact on the health-care system, social services, on families and communities, and quality of life. Significant efforts and progress have been made in learning the genetic causes of AMD, yet further research is needed to bring forth prevention and treatment strategies.
References
Seddon JM, Ajani UA, Mitchell BD : Familial aggregation of age-related maculopathy. Am J Ophthalmol 1997; 123: 199–206.
Meyers SM : A twin study on age-related macular degeneration. Trans Am Ophthalmol Soc 1994; 92: 775–843.
Jakobsdottir J, Conley YP, Weeks DE, Mah TS, Ferrell RE, Gorin MB : Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet 2005; 77: 389–407.
Fisher SA, Abecasis GR, Yashar BM et al: Meta-analysis of genome scans of age-related macular degeneration. Hum Mol Genet 2005; 14: 2257–2264.
Bansback N, Czoski-Murray C, Carlton J et al: Determinants of health related quality of life and health state utility in patients with age related macular degeneration: the association of contrast sensitivity and visual acuity. Qual Life Res 2007; 16: 533–543.
Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U : A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet 2006; 38: 1173–1177.
Edwards AO, Ritter III R, Abel KJ, Manning A, Panhuysen C, Farrer LA : Complement factor H polymorphism and age-related macular degeneration. Science 2005; 308: 421–424.
Klein RJ, Zeiss C, Chew EY et al: Complement factor H polymorphism in age-related macular degeneration. Science 2005; 308: 385–389.
Hageman GS, Anderson DH, Johnson LV et al: A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA 2005; 102: 7227–7232.
Haines JL, Hauser MA, Schmidt S et al: Complement factor H variant increases the risk of age-related macular degeneration. Science 2005; 308: 419–421.
Gold B, Merriam JE, Zernant J et al: Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet 2006; 38: 458–462.
Rivera A, Fisher SA, Fritsche LG et al: Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet 2005; 14: 3227–3236.
Hageman GS, Hancox LS, Taiber AJ et al: Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med 2006; 38: 592–604.
Age-Related Eye Disease Study Research Group: The Age-Related Eye Disease Study (AREDS): design implications. AREDS report no. 1. Control Clin Trials 1999; 20: 573–600.
Jacques PF : The potential preventive effects of vitamins for cataract and age-related macular degeneration. Int J Vitam Nutr Res 1999; 69: 198–205.
Age-Related Eye Disease Study Research Group: A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E and beta carotene for age-related cataract and vision loss: AREDS report no. 9. Arch Ophthalmol 2001; 119: 1439–1452.
Mitchell P, Foran S : Age-Related Eye Disease Study severity scale and simplified severity scale for age-related macular degeneration. Arch Ophthalmol 2005; 123: 1598–1599.
Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM : Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet 2007; 39: 1200–1201.
Allikmets R, Dean M : Bringing age-related macular degeneration into focus. Nat Genet 2008; 40: 820–821.
Hughes AE, Orr N, Patterson C et al: Neovascular age-related macular degeneration risk based on CFH, LOC387715/HTRA1, and smoking. PLoS Med 2007; 4: e355.
Schmidt S, Haines JL, Postel EA et al: Joint effects of smoking history and APOE genotypes in age-related macular degeneration. Mol Vis 2005; 11: 941–949.
Prosser BE, Johnson S, Roversi P et al: Structural basis for complement factor H linked age-related macular degeneration. J Exp Med 2007; 204: 2277–2283.
Fritsche LG, Loenhardt T, Janssen A et al: Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet 2008; 40: 892–896.
Schultz DW, Klein ML, Humpert A et al: Lack of an association of apolipoprotein E gene polymorphisms with familial age-related macular degeneration. Arch Ophthalmol 2003; 121: 679–683.
DeAngelis MM, Ji F, Kim IK et al: Cigarette smoking, CFH, APOE, ELOVL4, and risk of neovascular age-related macular degeneration. Arch Ophthalmol 2007; 125: 49–54.
Schmidt S, Klaver C, Saunders A et al: A pooled case–control study of the apolipoprotein E (APOE) gene in age-related maculopathy. Ophthalmic Genet 2002; 23: 209–223.
Yang Z, Stratton C, Francis PJ et al: Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med 2008; 359: 1456–1463.
Edwards AO, Chen D, Fridley BL et al: Toll-like receptor polymorphisms and age-related macular degeneration. Invest Ophthalmol Vis Sci 2008; 49: 1652–1659.
Allikmets A, Bergen AA, Dean M et al, the International AMD Genetics Consortium: Toll-like receptor 3 gene is not associated with geographic atrophy in age-related macular degeneration. NEJM 2009; (in press).
Cong R, Zhou B, Sun Q, Gu H, Tang N, Wang B : Smoking and the risk of age-related macular degeneration: a meta-analysis. Ann Epidemiol 2008; 18: 647–656.
Acknowledgements
This study was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and SAIC-Frederick under contract no. NO1-CO-12400 and with grants from the National Eye Institute EY13435 and EY017404 (RA); the Macula Vision Research Foundation; Wallach Foundation (RA); Elyachar Foundation (RA); Kaplen Foundation (RA); Wigdeon Point Charitable Foundation (RA); and an unrestricted grant to the Department of Ophthalmology, Columbia University, from Research to Prevent Blindness. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be marked as an advertisement in accordance with 18 USC Section 1734 solely to indicate this fact.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bergeron-Sawitzke, J., Gold, B., Olsh, A. et al. Multilocus analysis of age-related macular degeneration. Eur J Hum Genet 17, 1190–1199 (2009). https://doi.org/10.1038/ejhg.2009.23
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.23
Keywords
This article is cited by
-
Role of complement factor B rs4151667 (L9H) polymorphisms and its interactional role with CFH Y402H and C3 rs2230199 (R102G) risk variants in age-related macular degeneration: a case control study
BMC Ophthalmology (2020)
-
Association between genetic variation of complement C3 and the susceptibility to advanced age-related macular degeneration: a meta-analysis
BMC Ophthalmology (2018)
-
Genome-wide analyses identify common variants associated with macular telangiectasia type 2
Nature Genetics (2017)
-
Genotype distribution-based inference of collective effects in genome-wide association studies: insights to age-related macular degeneration disease mechanism
BMC Genomics (2016)
-
Genetic and environmental factors strongly influence risk, severity and progression of age-related macular degeneration
Signal Transduction and Targeted Therapy (2016)
