
Abstract
Mutations in the gene Indian Hedgehog (IHH) that cause Brachydactyly A-1 (BDA1) have been restricted to a specific region of the N-terminal active fragment of Indian Hedgehog involving codons 95, 100, 131, and 154. We describe two novel mutations in codons 128 and 130, not previously implicated in BDA1. Furthermore, we identified an independent mutation at codon 131 and we also describe a New Zealand family, which carries the ‘Farabee’ founder mutation and haplotype. All of the BDA1 mutations occur in a restricted area of the N-terminal active fragment of the IHH and are in contrast to those mutations causing an autosomal recessive acrocapitofemoral dysplasia, whose mutations are located at the distal N- and C-terminal regions of IHH-N and are physically separated from the BDA1-causing mutations. The identification of multiple independent mutations in codons 95, 100, and now in 131, implicate a discrete function for this region of the protein. Finally, we present a clinical review of all reported and confirmed cases of BDA1, highlighting features of the disorder, which add to the spectrum of the IHH mutations.
Similar content being viewed by others


Introduction
Brachydactyly A-1 (BDA1 MIM 112500) is characterized by shortness of all middle phalanges of the hands and toes, occasional terminal symphalangism, shortness of the proximal phalanges of the first digit, and short stature. Although BDA1 can occur as an isolated malformation,1, 2, 3, 4, 5, 6, 7 it has also been described as part of complex syndromes, with some of the most commonly reported associated disorders being nystagmus,8, 9 developmental delay, mental retardation,8, 9, 10 and scoliosis.8, 11, 12
BDA1 has the distinction of being the first disorder to be described as an autosomal dominant Mendelian trait in humans.13 Mutations in the Indian Hedgehog gene (IHH) were initially identified in three Chinese BDA1 families,14 and a second locus has been mapped to 5p13.3-p13.215 (MIM 607004). Furthermore, both the IHH and the chromosome 5p13.3-p13.2 region were excluded in at least one other BDA1-affected family, implicating no less than one additional locus in the development of BDA1.16 In addition to dominant mutations causing BDA1, recessive mutations in IHH cause acrocapitofemoral dysplasia (ACFD; MIM 607778).17 These patients presented with short stature, BDA1, and cone-shaped epiphyses of the tubular bones of the hands and the proximal end of the femur.17 The cone-shaped epiphyses appeared early in childhood and disappeared with premature fusion of the growth plate.
In the Ihh−/− mouse, the loss of Ihh signaling results in a limb reduction phenotype with a complete lack of osteoblast development in all bones that develop by endochondral ossification,18 highlighting the role played by Ihh in cartilage differentiation and in bone formation. Furthermore, an inversion of the sonic hedgehog locus has been shown to yield a murine brachydactyly phenotype in heterozygotes by a gain-of-function effect.19
In 1903, Farabee13described a large family from Pennsylvania with BDA1 and a few years later, three additional BDA1-affected families of English ancestry were described by Drinkwater.2, 3, 4 Descendants of Farabee's original family and two of Drinkwater's families were found to share a common IHH mutation resulting in a p.Asp100Asn amino acid substitution. In addition, these families shared a common haplotype flanking IHH, indicating that they share a common founder.20, 21
The only reported BDA1-causing mutation that was not restricted to codons 95, 100, and 131 of IHH is p.Thr154Ile.22 All four of these codons are highly conserved (Figure 1). Multiple mutations in codons 95 and 100 indicate that these codons may be mutational hot spots; moreover, their proximity to one another suggests that they may reside in a region of IHH that is of particular importance and is responsible for the normal functioning of the growth plate during bone development.

Alignment of the amino acid sequence of IHH with those of other species, as well as with human SHH and DHH. The amino acid sequence of the human IHH was aligned with those of mouse Ihh, chicken Ihh, African clawed frog (banded hedgehog), and zebrafish (echidna hedgehog). The alignment has been anchored to the Homo sapien IHH amino acid sequence, and the amino acid numbers are listed above. These were further aligned with the amino acid sequences of human SHH and DHH. Residues associated with disease are indicated by an arrow and by the wild-type amino acids in red. Heterozygous mutations D100N, R128Q, T130N, and E131K are associated with BDA1 in this study (denoted by an asterisk*). Other BDA1-causing heterozygous mutations E95K, D100E, and E131K were described by Gao et al14; D100N was described by McCready et al20, Giordano et al23, and McCready et al21; E95G was described by Kirkpatrick et al16; T154I was described by Liu et al22; and delE95 was described by Lodder et al7. Homozygous mutations, P46L and V190A, were described by Hellemans et al17 and are associated with ACFD.
Materials and methods
Four BDA1-affected families of diverse ethnic and regional backgrounds were studied. In all the cases, the disease was inherited as an autosomal dominant trait. Diagnosis was based on physical examination, radiographic findings when available, and family history. The study was approved by the Children's Hospital of Eastern Ontario Ethics Review Committee. After receiving informed consent, genomic DNA was extracted from peripheral venous blood or saliva samples using a QIAamp DNA blood mini-kit (Qiagen, Valencia, CA, USA) or an Oragene DNA self-collection kit (DNA Genotek, Ottawa, ON, Canada).
Sequence analysis
All three exons of IHH, including flanking splice sites and untranslated regions, were amplified by PCR and sequenced using primers and conditions described earlier.20 The single-exon gene, NOGGIN, was amplified and sequenced as described above. All primers and optimized conditions are described in Supplementary Table 1.
Restriction digest
To detect the c.383G>A or the c.389C>A nucleotide change in the IHH gene, exon 2 was amplified by PCR and subsequently digested, according to the manufacturer's instructions, with PstI or BstEII, respectively. Products were loaded on to a 1.5% agarose gel containing ethidium bromide, electrophoresed for 40 min at 100 V, and photographed under UV light. This procedure was repeated with 200 control DNA samples for both c.383G>A and c.389C>A.
Microsatellite markers
Seven markers from Marshfield's sex-averaged genetic map were examined (D2S2250, D2S433, D2S163, D2S1242, D2S424, D2S1323, and D2S126) along with two single nucleotide polymorphisms (SNPs) located upstream of exon 1 (rs437512, and rs1960326) and 3 SNPs in exon 3 (rs3731881, rs394452, and rs3099) of IHH. Genotyping was performed as described earlier.20
Results
Individuals from four families of diverse ethnic and regional backgrounds were examined for mutations within IHH. Although sporadic cases of BDA1 have been reported, all the families presented here show an autosomal dominant pattern of inheritance.
Family 1
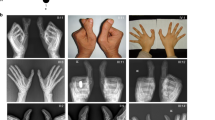
Four members of an American family with BDA1 segregating in at least four generations were examined. The family's ancestors are of German, Scottish, and Irish descent, with most of the migrants settling in southern United States. The family was ascertained, when an ultrasound revealed short limbs in a third-trimester fetus. The family was referred to the Stanford University Medical Center where the proband (individual 1-01 in Figure 2), the 43-year-old father of the fetus, was diagnosed with BDA1 on the basis of clinical and radiographic evaluations (Figure 3). The middle phalanges were very short, especially those in digits two and five. The proximal phalange of digit one was also quite short. The proband had short arms, but normal stature at a height of 5′10′′ (175 cm). Other phenotypic findings included limited dorsiflexion of the feet and tarsal coalition. The proband's father (individual 1-03 in Figure 2) and the father's sibling were also reportedly affected with BDA1. The father's sibling had an affected child, who in turn had three affected children. One of the children was reported to have a ‘problem with the palate’ that did not require repair. No further abnormalities were described in the family.

Pedigrees of four families with BDA1. Brachydactyly type A1 is transmitted as an autosomal dominant trait in all families. Probands are denoted by arrows. Numbers represent the sample number assigned to the DNA of individuals who participated in this study. Those members of family 4 above the dashed line represent members who were described earlier by Nissen.24

Hand radiographs of affected members of families 1, 3, and 4.
The presence of tarsal coalition in the proband drew attention to the candidate gene NOGGIN, but no polymorphisms or sequence variants were identified in the proband's DNA. However, sequencing of the IHH gene revealed a novel heterozygous c.383G>A nucleotide change in the DNA of individuals 1-01 and 1-03 (Figure 4a). Only the affected family members carried this nucleotide change, which resulted in a p.Arg128Gln amino acid substitution. The c.383G>A nucleotide change, which created a PstI restriction site, was not observed in 400 control chromosomes.

Mutations in IHH. (a) Family 1, c.383G>A; (b) Family 2, c.389C>A; (c) Family 3, c.391G>A; (d) Family 4, c.298G>A.
Family 2
Three members of a family of Indian descent with BDA1 segregating in at least three generations were examined. The proband (individual 2-01 in Figure 2) was referred to the University of Hong Kong's Queen Mary Hospital, where he was diagnosed with BDA1 based on clinical evaluations. Radiographs of the probands hands were not available. The middle phalanges were described as being very short, and images of the proband's flexed hands revealed that the middle phalanges in digits two and five were most likely missing or fused to the terminal phalange as only one interdigital joint was visible. This individual had a more severe form of BDA1 associated with distal symphalangism, scoliosis, and clubfoot. Interestingly, all the proband's blood relatives on his father's side were reported to have BDA1. DNA was obtained from the proband, his affected brother, and an affected cousin (individuals 2-02 and 2-03 in Figure 2).
A novel heterozygous c.389C>A nucleotide change was identified in the DNA of individual 2-01 (Figure 4b). To determine whether this change co-segregated with the malformation in this family, the DNA of all three available family members was screened by restriction digest with BstEII. Analysis revealed that all affected individuals carried the c.389C>A nucleotide change, which resulted in a p.Thr130Asn amino acid substitution. The c.389C>A nucleotide change was not observed in 400 control chromosomes.
Family 3
Three members of an Ashkenazi Jewish family residing in Israel with BDA1 reportedly segregating in at least four generations were examined. The family was ascertained, when the 34-year-old female proband (individual 3-01 in Figure 2) was referred to the Tel Aviv Sourasky Medical Center in Tel Aviv, Israel, for pre-implantation genetic diagnosis of BDA1 before undergoing in vitro fertilization. The proband was diagnosed with BDA1 based on clinical and radiological findings (Figure 3). The middle and distal phalanges were replaced with a single small chess-pawn-shaped bone in digits two to five, and she had very short proximal and distal phalanges in digit one. Her mother was also affected. No further abnormalities were described in this family. In the IHH, a heterozygous c.391G>A nucleotide change was observed in the DNA of individual 3-01 (Figure 4c). Sequence analysis revealed that only the affected individuals carried the nucleotide change, which resulted in a p.Glu131Lys amino acid substitution. As this nucleotide change has been earlier associated with BDA1 in a Chinese family,14 no control individuals were screened for this change.
Family 4
Seven members of a New Zealand family with BDA1 segregating as an autosomal dominant, fully penetrant disorder in at least eight generations were examined. The family's ancestors originated from England, with branches of the family settling in both Australia and New Zealand. The 35-year-old female proband (individual 4-01 in Figure 2) initially came to attention after a referral to the genetics clinic for evaluation of her short fingers. Radiographic analysis revealed absent middle phalanges in digits two to five in both the hands and feet, as well as shortened proximal phalanges in digit one (Figure 3). There was a single interphalangeal joint in each digit, and the proband could not bend her thumbs. Other clinical findings included syndactyly of the second and third toes, aching back and knees, hallux vulga, and absent lateral incisors. The proband's height was 163 cm (25th–50th percentile).
On examination at 59 years of age, the proband's affected mother (individual 4-02 in Figure 4) presented with pain in the lower back, knees, toes, and arches of feet. Other clinical features observed in the extended family included lumbar lordosis, extra teeth, and a shortened fifth metacarpal (Table 1). Within IHH, a heterozygous c.298G>A nucleotide change was present in the DNA of individuals 4-01 and 4-02. All the six affected family members were found to be carrying the c.298G>A change (Figure 4d), which resulted in a p.Asp100Asn amino acid substitution. As this mutation has been reported previously in the Farabee21 and Drinkwater20 pedigrees, no control individuals were screened. On the basis of reports that the family's ancestors were from England, a possible association of the New Zealand kindred with the Drinkwater and Farabee families was addressed by evaluating whether the affected members carried the same ancestral haplotype. Seven polymorphic markers and five SNPs spanning a 4.82-cM region flanking the IHH gene were studied. When compared with the DNA of the Drinkwater and Farabee families, a common shared haplotype was observed between markers D2S2250 and D2S1323 (data not shown). Three synonymous exonic polymorphisms were detected in the sequence, all of which were present in the NCBI SNP database (Build 129).
Discussion
Indian Hedgehog is best known for its role in mediating condensation, growth and differentiation of long bone cartilage templates,27 and mutations in the gene are known to cause the dominant and recessive disorders, BDA1 and ACFD, respectively. We have investigated the possibility that mutations in IHH were responsible for BDA1 in four families of diverse geographical and ethnic origins presenting with varying phenotypes.
We identified a novel heterozygous c.383G>A mutation in family 1. This nucleotide change causes a novel p.Arg128Gln amino acid substitution. Individual 1-01 presented with a complex BDA1 phenotype that included short arms, tall stature, tarsal coalition, and limited dorsiflexion, none of which were seen in the other three families described here. Tall stature with BDA1 has been described earlier (Table 1).11 Mesomelic shortening of the limbs has also been described in a family with the Osebold–Remondini Syndrome, also referred to as Brachydactyly type A6 (MIM 112910).28 Affected individuals in the family had small or absent middle phalanges, radial deviation of index fingers, and abnormal carpal and tarsal bones. In addition to investigating IHH, the candidate gene NOGGIN, was excluded in this family. Heterozygous missense and nonsense NOGGIN mutations have been found to cause multiple synostoses syndrome, proximal symphalangism, and carpal–tarsal coalition syndrome, all of which present clinically with symphalangisms and/or carpal–tarsal fusions.29, 30, 31, 32 A phenotype of BDA1, normal stature, and short arms seen with a novel p.Arg128Gln mutation indicates that the BDA1 phenotype associated with the IHH mutations may not be restricted to the middle phalanx.
We also identified a novel heterozygous c.389C>A mutation in family 2, a family of Indian descent. This mutation co-segregated with the BDA1 phenotype in the family, and causes a novel p.Thr130Asn amino acid substitution. However, the proband also presented with distal symphalangism, scoliosis, and clubfoot. Interestingly, a heterozygous c.391G>A mutation was identified in family 3, a family of Ashkenazi Jewish descent residing in Israel. An identical nucleotide change has been reported previously in a Miao Chinese kindred affected with BDA1.14 The nucleotide change causes a p.Glu131Lys amino acid substitution in a residue that is highly conserved in many hedgehog proteins (Figure 1). As these two families hail from different regional and ethnic backgrounds, it seems unlikely that this mutation is the result of a common ancestor. Rather, it most likely occurred independently in both families, indicating that c.389C>A may represent a mutational hot spot in IHH. Affected individuals of the Chinese family were reported to be missing the middle phalanges in digits two to five, and radiographs show the presence of the same chess-pawn-shaped distal bone observed in the affected individuals of the Israeli family. This phenotype, which is common to both families, suggests a particularly important role for Glu131 in the IHH function throughout skeletal development.
In 1933, Nissen24 reported a BDA1 family that emigrated from England to Australia around 1840, with one branch subsequently migrating to New Zealand around 1850. Members of this branch were examined and found to have characteristic deformities similar to that of the Drinkwater and Farabee families.24 The proband's aunt (family 4) was described at the age of 5 years by Nissen, establishing that the remaining individuals examined here are descendants of that family.24 Interestingly, affected members of the family presented with a remarkable phenotypic heterogeneity. All the six affected members who were examined were shorter in stature than their unaffected siblings. Radiographs were not available for all members, but the degree of shortening of the middle phalanges and the presence or absence of distal symphalangism presumably account for some of the phenotypic heterogeneity observed in the family. Four of the six affected individuals who were examined had 2–3 syndactyly of the toes. The affected individuals 4-01 and 4-02 were missing lateral incisors, whereas the affected individual 4-03 was reported to have extra teeth. Supernumerary teeth and dental anomalies have been described earlier in conjunction with Brachydactyly types B and E, angel-shaped phalangoepiphyseal dysplasia, and autosomal recessive and dominant Robinow's syndromes,33, 34, 35, 36 but have not been associated with BDA1 to date. Interestingly, sonic hedgehog and its downstream targets, Ptch1 and Gli1, have been clearly implicated in both murine and fish tooth development;25, 26 however, no role for IHH has been delinated. Family 4 was found to carry the historic c.298G>A (p.D100N) mutation, as well as the haplotype flanking the IHH gene common to the two Drinkwater families and to the Farabee family, indicating that these families share a common founder.20, 21 This founder mutation is speculated to have originated at least 12 generations ago. To date, three other BDA1-affected families of Italian, American, and Chinese descent have been found to share this same IHH mutation.21, 23, 37 Although it remains possible that the families of Italian and American descent may have originated from this same common founder, the Chinese mutation was found to be flanked by a different haplotype.37 Another mutation affecting the same codon, c.300C>A, has also been associated with BDA1 in another Chinese family.14 This nucleotide change caused a p.Asp100Glu amino acid substitution. The existence of at least three independent mutations in this codon suggests that Asp100 may also represent a mutational hot spot.
With the exception of p.delE95,7 all the BDA1-causing IHH mutations are missense and are limited to a 59 amino-acid region of the N-terminal active fragment (IHH-N) spaning codons 95–154. Including the two novel mutations described here, p.Arg128Gln and p.Thr130Asn, the three-dimensional structure of the IHH-N reveals that all the BDA1-causing IHH mutations are restricted to the central region of IHH-N (Figure 5). In addition, the limited number of codons involved in the disease, borne out by multiple independent mutations in codons 95, 100, and now in 131, suggests a discrete function for this region of the protein. This is in contrast to the IHH mutations known to cause autosomal recessive ACFD, which are located at the distal N- and C-terminal regions of the IHH-N and are physically separated from the BDA1-causing IHH mutations (Figure 5). A phenotype of BDA1, with average stature and short arms seen with a novel p.Arg128Gln mutation, indicates that the phenotype may not be restricted to the middle phalanx. In a review of the published cases of BDA1 with IHH mutations (see Table 1), shortening of the middle phalanges of the hands was the mildest phenotype. In one family, the feet were reported to be normal; however, lower limb X-rays were not provided in the paper.22 The involvement of the phalanges and metacarpals (metatarsals) is quite variable even within the same family. Shortening of the first metacarpal, which is typically a distinguishing feature of Brachydactyly type C is a rare but reported finding.37 Generalized musculoskeletal complaints including arthritis were often reported, as well as the more specific findings of clubfoot and scoliosis. The family described by Raff et al,12 has not had a mutation reported to date, but the X-rays and clinical photos of the hands clearly show the presence of BDA1. Their family has associated abnormal menisci and scoliosis that had been well documented. This may provide an insight into the issues in some of the other families. In the Dutch family affected with ACFD,17 the carrier parents were noted to have phalangeal shortening when measured formally. The pattern would be consistent with a mild BDA1. Although short stature has been used in the definition of BDA1, in reviewing the reported cases, this is clearly not always the case. Farabee had noted the short stature in his initial paper; however in later generations, the height of affected family members is unremarkable in comparison with their unaffected relatives.

Three-dimensional reconstruction of the N-terminal active fragment of Indian Hedgehog defining the positions of the amino acids, whose mutation have been implicated in Brachydactyly type A1 or Acrocapitofemoral dysplasia pathogenesis. To compare the locations of the Indian Hedgehog mutations, the crystal structure of the amino-terminal domain of mouse Shh (1VHH.pdb) was used because of its high similarity to IHH. The equivalent positions were utilized. Numbers indicate the amino acid positions. ACFD mutations (blue); BDA1 mutations at codon 100 (red), codons 95, 131, and 154 (green), and codons 128, 130 (orange) are shown. N and C termini are indicated. The ribbons represent α-helices. The three representations are shown from a perspective, in which the N and C termini are closest to the viewer; at the top of the molecule; and furthest away from the viewer, projecting into the page. All the BDA1 mutations appear to cluster in the central portion of IHH-N.
References
Armour CM, Bulman DE, Hunter AG : Clinical and radiological assessment of a family with mild brachydactyly type A1: the usefulness of metacarpophalangeal profiles. J Med Genet 2000; 37: 292–296.
Drinkwater H : An account of a brachydactylous family. Proc R Soc Edinb 1908; 28: 35–57.
Drinkwater H : Account of a family showing minor brachydactyly. J Genet 1912; 2: 21–40.
Drinkwater H : A second brachydactylous family. J Genet 1915; 4: 323–339.
Haws DV, McKusick VA : Farabee's brachydactylous kindred revisited. Bull Johns Hopkins Hosp 1963; 113: 20–30.
Yang X, She C, Guo J et al: A locus for brachydactyly type A-1 maps to chromosome 2q35–q36. Am J Hum Genet 2000; 66: 892–903.
Lodder EM, Hoogeboom AJ, Coert JH, de Graaff E : Deletion of 1 amino acid in Indian hedgehog leads to brachydactylyA1. Am J Med Genet A 2008; 146A: 2152–2154.
Slavotinek A, Donnai D : A boy with severe manifestations of type A1 brachydactyly. Clin Dysmorphol 1998; 7: 21–27.
Tsukahara M, Azuno Y, Kajii T : Type A1 brachydactyly, dwarfism, ptosis, mixed partial hearing loss, microcephaly, and mental retardation. Am J Med Genet 1989; 33: 7–9.
Grange DK, Balfour IC, Chen SC, Wood EG : Familial syndrome of progressive arterial occlusive disease consistent with fibromuscular dysplasia, hypertension, congenital cardiac defects, bone fragility, brachysyndactyly, and learning disabilities. Am J Med Genet 1998; 75: 469–480.
Sillence DO : Brachydactyly, distal symphalangism, scoliosis, tall stature, and club feet: a new syndrome. J Med Genet 1978; 15: 208–211.
Raff ML, Leppig KA, Rutledge JC, Weinberger E, Pagon RA : Brachydactyly type A1 with abnormal menisci and scoliosis in three generations. Clin Dysmorphol 1998; 7: 29–34.
Farabee WC : Hereditary and Sexual Influences in Meristic Variation: A Study of Digital Malformations in Man. Boston: Harvard University, 1903.
Gao B, Guo J, She C et al: Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat Genet 2001; 28: 386–388.
Armour CM, McCready ME, Baig A, Hunter AG, Bulman DE : A novel locus for brachydactyly type A1 on chromosome 5p13.3-p13.2. J Med Genet 2002; 39: 186–188.
Kirkpatrick TJ, Au KS, Mastrobattista JM, McCready ME, Bulman DE, Northrup H : Identification of a mutation in the Indian Hedgehog (IHH) gene causing brachydactyly type A1 and evidence for a third locus. J Med Genet 2003; 40: 42–44.
Hellemans J, Coucke PJ, Giedion A et al: Homozygous mutations in IHH cause acrocapitofemoral dysplasia, an autosomal recessive disorder with cone-shaped epiphyses in hands and hips. Am J Hum Genet 2003; 72: 1040–1046.
St-Jacques B, Hammerschmidt M, McMahon AP : Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 1999; 13: 2072–2086.
Niedermaier M, Schwabe GC, Fees S et al: An inversion involving the mouse Shh locus results in brachydactyly through dysregulation of Shh expression. J Clin Invest 2005; 115: 900–909.
McCready ME, Sweeney E, Fryer AE et al: A novel mutation in the IHH gene causes brachydactyly type A1: a 95-year-old mystery resolved. Hum Genet 2002; 111: 368–375.
McCready ME, Grimsey A, Styer T, Nikkel SM, Bulman DE : A century later Farabee has his mutation. Hum Genet 2005; 117: 285–287.
Liu M, Wang X, Cai Z et al: A novel heterozygous mutation in the Indian hedgehog gene (IHH) is associated with brachydactyly type A1 in a Chinese family. J Hum Genet 2006; 51: 727–731.
Giordano N, Gennari L, Bruttini M et al: Mild brachydactyly type A1 maps to chromosome 2q35–q36 and is caused by a novel IHH mutation in a three generation family. J Med Genet 2003; 40: 132–135.
Nissen KI : A study in inherited brachydactyly. Ann Eugen 1933; 5: 281–301.
Fraser GJ, Bloomquist RF, Streelman JT : A periodic pattern generator for dental diversity. BMC Biol 2008; 6: 32.
Cobourne MT, Miletich I, Sharpe PT : Restriction of sonic hedgehog signalling during early tooth development. Development 2004; 131: 2875–2885.
Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ : Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996; 273: 613–622.
Osebold WR, Remondini DJ, Lester EL, Spranger JW, Opitz JM : An autosomal dominant syndrome of short stature with mesomelic shortness of limbs, abnormal carpal and tarsal bones, hypoplastic middle phalanges, and bipartite calcanei. Am J Med Genet 1985; 22: 791–809.
Dixon ME, Armstrong P, Stevens DB, Bamshad M : Identical mutations in NOG can cause either tarsal/carpal coalition syndrome or proximal symphalangism. Genet Med 2001; 3: 349–353.
Gong Y, Chitayat D, Kerr B et al: Brachydactyly type B: clinical description, genetic mapping to chromosome 9q, and evidence for a shared ancestral mutation. Am J Hum Genet 1999; 64: 570–577.
Mangino M, Flex E, Digilio MC, Giannotti A, Dallapiccola B : Identification of a novel NOG gene mutation (P35S) in an Italian family with symphalangism. Hum Mutat 2002; 19: 308.
Takahashi T, Takahashi I, Komatsu M et al: Mutations of the NOG gene in individuals with proximal symphalangism and multiple synostosis syndrome. Clin Genet 2001; 60: 447–451.
Gorlin RJ, Sedano HO, Odont : Cryptodontic brachymetacarpalia. Birth Defects Orig Artic Ser 1971; 7: 200–203.
Holder-Espinasse M, Escande F, Mayrargue E et al: Angel shaped phalangeal dysplasia, hip dysplasia, and positional teeth abnormalities are part of the brachydactyly C spectrum associated with CDMP-1 mutations. J Med Genet 2004; 41: e78.
Hunter AG, McAlpine PJ, Rudd NL, Fraser FC : A ‘new’ syndrome of mental retardation with characteristic facies and brachyphalangy. J Med Genet 1977; 14: 430–437.
Robinow M, Silverman FN, Smith HD : A newly recognized dwarfing syndrome. Am J Dis Child 1969; 117: 645–651.
Zhu G, Ke X, Liu Q et al: Recurrence of the D100N mutation in a Chinese family with brachydactyly type A1: evidence for a mutational hot spot in the Indian hedgehog gene. Am J Med Genet A 2007; 143: 1246–1248.
Acknowledgements
The authors thank the families for their participation and Dr John Christodoulou for his help in arranging the examination of two patients. This work was funded by the Canadian Institute of Health Research (DEB). LR is funded by an Ontario Graduate Studentship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Byrnes, A., Racacho, L., Grimsey, A. et al. Brachydactyly A-1 mutations restricted to the central region of the N-terminal active fragment of Indian Hedgehog. Eur J Hum Genet 17, 1112–1120 (2009). https://doi.org/10.1038/ejhg.2009.18
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.18
Keywords
This article is cited by
-
Suppression of apoptosis impairs phalangeal joint formation in the pathogenesis of brachydactyly type A1
Nature Communications (2024)
-
Inactivation of Ihh in Sp7-Expressing Cells Inhibits Osteoblast Proliferation, Differentiation, and Bone Formation, Resulting in a Dwarfism Phenotype with Severe Skeletal Dysplasia in Mice
Calcified Tissue International (2022)
-
Deletion of 2 amino acids in IHH in a Japanese family with brachydactyly type A1
BMC Medical Genomics (2021)
-
Altered microRNAs in C3H10T1/2 cells induced by p.E95K mutant IHH signaling
Hereditas (2021)
-
A novel variant of IHH in a Chinese family with brachydactyly type 1
BMC Medical Genetics (2020)
