
Abstract
In this study we clinically and genetically characterize a consanguineous family with a homozygous novel missense mutation in the δ-sarcoglycan gene and a second δ-sarcoglycan mutation that has previously been reported to cause severe autosomal-dominant dilated cardiomyopathy. We identified a novel missense mutation in exon 6 (p.A131P) of the δ-sarcoglycan gene, which in a homozygous state leads to the clinical picture of a limb girdle muscular dystrophy. In four heterozygous carriers for the mutation, aged 3–64 years, a second sequence variant in exon 6 (p.S151A) of the δ-sarcoglycan gene was detected on the other allele. This second missense change had previously been reported to be responsible for fatal autosomal-dominant dilated cardiomyopathy at young age. Comprehensive clinical and cardiac investigation in all of the compound heterozygous family members revealed no signs of cardiomyopathy or limb girdle muscular dystrophy. Our findings demonstrate that, even in the presence of a second disease-causing mutation, the p.S151A mutation in the δ-sarcoglycan gene does not result in cardiomyopathy. This finding questions the pathological relevance of this sequence variant for causing familial autosomal-dominant dilated cardiomyopathy and thereby the role of the δ-sarcoglycan gene in general as a disease-causing gene for autosomal-dominant dilated cardiomyopathy.
Similar content being viewed by others

Introduction
The sarcoglycans are part of the dystrophin–glycoprotein complex (DGC), an oligomeric complex spanning the plasma membrane of skeletal and cardiac muscle fibres. It is assumed that integrity of the sarcoglycan complex is crucial to maintain the mechanical and non-mechanical linkage between the subsarcolemmal cytoskeleton and the extracellular matrix.1, 2, 3, 4 Six sarcoglycans, α-, β-, γ-, δ-, ɛ- and ζ-sarcoglycan, have so far been identified. Mutations in the genes for α-, β-, γ- and δ-sarcoglycan (SGCA; B, G and D) cause a heterogeneous group of autosomal recessive limb girdle muscular dystrophies (LGMD2C-F).5, 6, 7 The sarcoglycan-deficient LGMDs are characterized by progressive weakness of the pelvic and shoulder girdle musculature with symptoms ranging from very severe to mild.8, 9, 10 Patients with LGMD2C-F, in particular with β-sarcoglycan-deficient LGMD2E and δ-sarcoglycan-deficient LGMD2F, often develop a progressive and potentially fatal dilated cardiomyopathy (DCM) in association with muscular dystrophy.11 Mutations in the ɛ-sarcoglycan gene are associated with a dominant form of myoclonus dystonia syndrome,12 whereas so far no disease has been associated with mutations in the ζ-sarcoglycan gene. Dominant inheritance has not been reported in sarcoglycan-deficient LGMD. However, a previous report described autosomal-dominant mutations in the δ-sarcoglycan (SGCD) gene in patients with familial and sporadic cases of DCM without significant involvement of the skeletal muscle.13
Generally, DCM is a myocardial disease characterized by left ventricular dilatation and impaired contraction of the left ventricle and a 5-year mortality rate estimated up to 50% after onset of symptoms.14, 15 The underlying causes for DCM are heterogeneous but the disease is familial in approximately 20–35% of cases, typically showing an autosomal-dominant mode of inheritance.16, 17, 18, 19 Recently candidate gene screenings and linkage analyses in large families have successfully been applied for the identification of many disease genes including the SGCD gene.20 So far three putative DCM-associated mutations in the SGCD gene have been described whereas only one of them (p.S151A) has been reported to cause familial autosomal-dominant DCM.13, 21 However, the prevalence of mutations in the SGCD gene responsible for DCM was estimated to be less than one percent and it still remains controversial whether the SGCD gene is a predominant disease-causing gene in patients with DCM.21, 22
Here we characterized a large consanguineous family with LGMD2F caused by a novel homozygous missense mutation in exon 6 of the SGCD gene. In the same family, four heterozygous carriers for the mutation carried the sequence variant p.S151A on the other δ-sarcoglycan allele that had previously been reported to cause familial autosomal-dominant DCM.13 In the report from Tsubata et al,13 three patients carrying the p.S151A sequence variant developed DCM with heart failure. Two of the patients died of sudden cardiac death at an early age (17–37 years) and the third patient required heart transplantation at the age of 21 years.
In our family, the p.S151A sequence variant showed no pathological relevance for cardiac disease. Comprehensive clinical examination of seven family members carrying the p.S151A sequence variant did not reveal any clinical signs of cardiomyopathy. The existence of δ-sarcoglycan associated autosomal-dominant DCM, therefore, remains controversial.
Methods
Patients
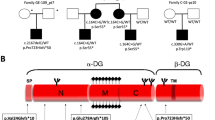
The family pedigree is depicted in Figure 1. The index patient (pedigree no. IV:11; 9-year-old girl) and two younger siblings (pedigree no. IV:12 and 13; 7-year-old girl and 5-year-old boy, respectively) with LGMD are three of eight children of consanguineous parents of Arabic origin (pedigree no. III:10; 31-year-old father and III:14; 35-year-old mother). The non-affected parents are first degree cousins with four siblings on the father's side (pedigree no. III:3, 5, 7, 9) and six siblings on the mother's side (pedigree no. III:15–17). Genetic analysis was performed in 19 family members. None of the other family members showed any signs of muscular dystrophy, and there is no family history of neuromuscular diseases or cardiac disease.

Pedigree: Black filled circles and squares represent LGMD2F affected females and males, respectively.
The three affected patients underwent clinical examinations, including manual muscle testing (MRC scale), blood tests for creatine kinase (CK), lung function tests and cardiac assessments.
Cardiac assessments
Standard 12-lead-ECGs and echocardiograms were performed in all the three patients, their father (pedigree no. III:10) and a paternal uncle (pedigree no. III:7; 31 years old). ECGs were examined for features suggestive of cardiomyopathy – P-wave morphology and QRS-voltage criteria for atrial or ventricular hypertrophy, respectively, presence of repolarization abnormalities or pathological Q-waves. Heart function was assessed by transthoracic echocardiography and tissue Doppler assessment. Image sets were obtained in standard parasternal long and short axes and apical four-chamber views. Left ventricular dimensions and systolic function were measured by fractional shortening, derived from M-mode chamber dimensions, and ejection fraction derived from a 16-segment wall motion scoring method. Right ventricular systolic function was assessed qualitatively. Systolic function was also assessed by tissue-Doppler measurements of four ventricular segments (lateral, septal, anterior, inferior) at the mitral valve ring. Diastolic ventricular function was assessed using the E/E′ ratio of the mitral flow method. All heart valves were assessed by 2-D inspection and Doppler flow measurements for the presence of structural or functional abnormalities.
Muscle biopsy
A muscle biopsy was obtained from the 6-year-old index patient (pedigree no. IV:11). Samples were mounted on cork, transversely orientated and rapidly frozen in isopentane. Cryostat sections were stained according to established methods.
Using a DAKO Autostainer plus, serial sections were immunostained with three different monoclonal antibodies to dystrophin specific for the rod-domain (clone Dy4/6D3, dilution 1:30; Novocastra), C-terminus (clone Dy8/6C5, dilution 1:50; Novocastra), and N-terminus (clone Dy10/12B2, dilution 1:15; Novocastra), α-(cloneAd1/20A6, dilution 1:100; Novocastra), β-(clone Sarc/5B1, dilution 1:50; Novocastra), γ-(clone 35DAG/21B5, dilution 1:300; Novocastra), and δ-sarcoglycan (clone Sarc3/12C1, dilution 1:100; Novocastra). Immunoreactions were visualized with Histofine® Simple Stain MAX-PO (Multi; Nichirei Biosciences).
Mutation screening
DNA was extracted from peripheral blood samples according to the standard protocols following patients' informed consent. In the index patient screening for mutations was performed by DHPLC (denaturing high performance liquid chromatography; Transgenomic, Cheshire, UK; analysis software Wavemaker 4.1) for all exons of the sarcoglycan (SGC) genes (SGCA, 9 exons; SGCB, 6 exons; SGCD, 8 exons). DNAs of the exons with conspicuous melting curves were sequenced (MegaBace1000; Amersham). The SGCG gene (8 exons) was sequenced directly. To exclude a large deletion of one allele of the SGCD gene two-colour multiplex ligation-dependent probe amplification (MLPA; Ceq8000; Beckman Coulter) was applied.
The mutations in the other family members were detected using direct bidirectional fluorescent sequencing of exon 6 of the SGCD gene. Sequences were analysed using DNASTAR SeqMAN 5.05 and Mutation Surveyor v3.10 semi-automated analysis software (SoftGenetics, USA).
Results
Clinical features and muscle biopsy findings
Three children (pedigree no. IV:11, 12, 13) of consanguineous parents were referred to the neuromuscular outpatient clinic. All three showed elevated serum CK levels (IV11: 8686 U/l; IV12: 25431 U/l and IV13: 20325 U/l), a waddling gait with difficulties to climb stairs, calf hypertrophy and a Gowers' manoeuvre. The two older siblings (pedigree no. IV:11, 12) showed pronounced muscle weakness in the pelvic and shoulder girdle muscles (MRC power grade 3− to 3+), whereas the youngest sibling was only mildly affected (pedigree no. IV:13). In all three patients, lung function testing did not reveal any signs of respiratory weakness with normal values for forced expiratory volume and vital capacity and the peak expiratory flow.
Histopathology of a muscle biopsy from the index patient obtained at the age of 6 years showed an extensive and active dystrophic process (Figure 2a). Immunohistochemical analysis detected a complete absence of α-, β-, γ- and δ-sarcoglycan in the presence of dystrophin, suggesting the diagnosis of a sarcoglycanopathy (Figure 2b–f). Figure 2f shows diffuse intracellular staining for δ-sarcoglycan indicating cytoplasmic accumulation of defective protein.

Histopathological (H&E) and immunohistochemical analysis of the dystrophin–glycoprotein complex in the muscle biopsy from the LGMD2F index patient (200-fold original magnification) demonstrate an active dystrophic process (a) and a virtual absence of α-, β-, γ- and δ-sarcoglycan (c–f) in the presence of dystrophin (b).
The patients underwent cardiac assessments, which showed no abnormalities. There was no evidence for either global or segmental left ventricular dysfunction or hypertrophy on echocardiographic or tissue-Doppler investigations. Left ventricular end-systolic and end-diastolic dimensions were within normal limits and 12-lead ECG recordings were all normal.
Genetic analysis
Identification of a novel δ-sarcoglycan mutation
All sarcoglycan genes were sequenced in the index patient (pedigree no. IV:11) and a single nucleotide substitution at position 391 (G for C), leading to an amino-acid change at codon 131 from alanine to proline, was detected in exon 6 of the SGCD gene. This sequence variant was subsequently identified in all the three affected patients in a homozygous state. Both parents were heterozygous carriers for the mutation. MLPA of the SGCD gene was also performed in the index patient and no deletion or duplication was detected.
The carrier status for the novel p.A131P mutation was tested in the extended family as consanguineous partnerships were planned. Direct sequence analysis of the coding region of exon 6 of the SGCD gene was performed in 19 relatives of the affected children (pedigree; Figure 1). Besides the parents (pedigree no. III:10 and III:14) of the three patients, another eight family members (pedigree no. II:5, III:7, IV:5, IV:6, IV:7, IV:8, IV:9 and IV:14) were found to carry the sequence variant p.A131P in a heterozygous state. In accordance with the autosomal recessive mode of inheritance none of the carriers showed any clinical signs of skeletal muscle weakness.
Sequence variant reported to cause fatal autosomal-dominant DCM
In addition to the novel disease-causing p.A131P mutation, direct sequence analysis of exon 6 in the father of the index patient (pedigree no. III:10) revealed a second heterozygous mutation in the SGCD gene. This sequence variant was not identified in the three affected children and is therefore located on the paternal allele that does not carry the disease-causing mutation.
The second sequence variant, a substitution of a single nucleotide (T for G) resulting in an amino-acid change from serine to alanine at codon 151 (p.S151A), had previously been described to cause fatal autosomal-dominant DCM.13 The same heterozygous mutation, p.S151A, was also identified in six other family members from 3 to 64 years of age (pedigree no. II:4, III7, IV:4, IV:9, IV:10, IV:14 ). Besides the father of the index patient (pedigree no. III:10), three other family members were carrying both the sequence variant p.A131P and p.S151A in a compound heterozygote manner (pedigree no. III:7; 32 years; IV:9; 11 years and IV:14; 3 years). Skeletal muscle and cardiac assessment performed in the compound heterozygote family members revealed no clinical signs of LGMD2F or DCM. ECG and echocardiography showed normal systolic and diastolic pump function and ventricular dimensions. There was no family history of muscle weakness, sudden death or heart disease. There were no further polymorphisms detected in the SGCD gene in addition to the p.S151A and p.A131P sequence variants in this family. In the index patient, four additional polymorphisms were found in the SGCG gene whereas no sequence variants were found in the SGCA or SGCB gene.
Discussion
The autosomal recessive sarcoglycan-deficient LGMD2C-F are a heterogeneous group of progressive muscle diseases associated with DCM.11 Similar to the animal models for δ-sarcoglycan-deficiency, the cardiomyopathic hamster models and the δ-sarcoglycan knock-out mouse, patients can already present severe cardiomyopathy at young age in addition to muscular dystrophy.23, 24 Cardiomyopathy is not a core component of LGMD2F and there have been reports of several patients without cardiac involvement.25 On the other hand, it has been suggested that mutations in the SGCD gene may cause autosomal-dominant DCM independently from symptoms of LGMD.13
Here we characterize a consanguineous family that has been diagnosed with LGMD2F caused by a novel homozygous missense mutation (p.A131P) in the SGCD gene. In the same family we found a sequence variant (p.S151A) in the SGCD gene that had previously been associated with fatal autosomal-dominant DCM.13 Neither the p.S151A carriers nor the compound heterozygous carriers for the two mutations showed any signs of heart disease, which calls into question the pathological relevance of the p.S151A mutation.
Mutations in the SGCD gene have been reported to cause familial autosomal-dominant DCM.13 In the study from Tsubata et al, 50 young DCM patients between 3 days and 18 years of age and one DCM family were screened for mutations in the SGCD gene, and two sequence variants were found. The sequence variant p.S151A was detected in three family members, who all showed symptoms of DCM, and a second sequence variant in position 238 (ΔK238) was found in two sporadic cases. No patient had signs of skeletal muscle disease. Tsubata et al concluded that the p.S151A sequence variant caused a fatal form of autosomal-dominant DCM characterized by heart failure, need for heart transplantation and sudden cardiac death at very young age. The authors hypothesized that DCM is mainly a cytoskeletopathy, which can be caused by disruption of the DGC and that δ-sarcoglycan is the disease-causing gene in their patients. Based on this report the existence of autosomal-dominant DCM caused by mutations in the SGCD gene was postulated.
In our study, besides a novel autosomal recessive mutation (p.A131P) identified to cause LGMD2F, the previously described p.S151A mutation was found in several family members of a large consanguineous family. According to our findings, there is no evidence that the p.S151A sequence variant causes DCM or LGMD in this family. The mutation was detected in seven family members between 3 and 64 years of age, and four of them were compound heterozygotes, carrying the LGMD-causing p.A131P mutation and the p.S151A sequence variant.
None of the examined carriers had a history or showed clinical signs of DCM or LGMD, and all cardiac examinations performed were normal. Even the oldest family member carrying the mutation (pedigree no. II:4; 64 years old) did not show any clinical signs of DCM. However, we cannot exclude the possibility that the identical mutation in the SGCD gene may lead to a milder, subclinical cardiac phenotype, as previously shown for another δ-sarcoglycan mutation (p.A71T) in a small Finnish family with DCM.21 In this family, patients were considerably older than those described by Tsubata et al and phenotypes have been reported to be less severe with a good response to medication. However, only two mutation carriers were found and definitive conclusions could not be presented. Thus, the authors reasoned that defects in the SGCD gene seem to be rare in patients with DCM. In our family, we have no information about cardiac expression levels of the allele, and effects of modifier genes on the penetrance in patients carrying the p.S151A sequence variant cannot be excluded. For instance, Gouveia et al reported a case of mild phenotype in a LGMD2F family with a strong SGCD mutation due to a retention of the other three sarcoglycans in the sarcolemma.26 It has also been speculated that the compensatory overexpression of ζ-sarcoglycan may lead to a milder phenotype in LGMD2F patients although this has so far not been proven.26 In wild-type mouse, cardiac muscle ζ-sarcoglycan is only weakly expressed.27
Although the p.S151A mutation was not detected in 200 healthy controls in the report by Tsubata et al, the possibility remains that this mutation is a rare non-pathological polymorphism. Studies to confirm an effect of this sequence variant on the protein function have not been performed. Transgenic mice overexpressing the p.S151A sequence variant develop DCM at a young age with enhanced lethality, despite the fact that convincing data in patients are still lacking.28 However, transgenic mice overexpressing a normal SGCG gene also develop myopathy and cardiomyopathy.29 Heydemann et al found up to sevenfold overexpression of the S151A sequence variant in hearts of their mice, which may suggest that rather the overall level of gene expression than the expression of the sequence variant has been responsible for the pathology in mice.28 The other sequence variant (ΔK238) in the SGCD gene described by Tsubata et al was found in two patients with sporadic DCM but in the absence of clear functional data and cosegregation with the phenotype in an autosomal-dominant manner no final conclusion can be drawn.
In conclusion, carriers of the p.S151A mutation in the δ-sarcoglycan gene, previously described to cause autosomal-dominant DCM, showed no evidence of cardiac pathology, despite the fact that several of them carried an additional pathogenic mutation on the other allele. This finding calls into question the pathological relevance of this p.S151A sequence variant for causing familial autosomal-dominant DCM and thereby the existence of an autosomal-dominant mode of inheritance for the pathogenic mutations in the SGCD gene.
References
Cohn RD, Campbell KP : Molecular basis of muscular dystrophies. Muscle Nerve 2000; 23: 1456–1471.
Hack AA, Lam MY, Cordier L et al: Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J Cell Sci 2000; 113: 2535–2544.
Heydemann A, McNally EM : Consequences of disrupting the dystrophin-sarcoglycan complex in cardiac and skeletal myopathy. Trends Cardiovasc Med 2007; 17: 55–59.
Ozawa E, Mizuno Y, Hagiwara Y, Sasaoka T, Yoshida M : Molecular and cell biology of the sarcoglycan complex. Muscle Nerve 2005; 32: 563–576.
Nigro V, de Sa Moreira E, Piluso G et al: Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta-sarcoglycan gene. Nat Genet 1996; 14: 195–198.
Noguchi S, McNally EM, Ben Othmane K et al: Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science 1995; 270: 819–822.
Bonnemann CG, Modi R, Noguchi S et al: Beta-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet 1995; 11: 266–273.
Zatz M, de Paula F, Starling A, Vainzof M : The 10 autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord 2003; 13: 532–544.
Zatz M, Vainzof M, Passos-Bueno MR : Limb-girdle muscular dystrophy: one gene with different phenotypes, one phenotype with different genes. Curr Opin Neurol 2000; 13: 511–517.
Bushby KM : The limb-girdle muscular dystrophies-multiple genes, multiple mechanisms. Hum Mol Genet 1999; 8: 1875–1882.
Politano L, Nigro V, Passamano L et al: Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromuscul Disord 2001; 11: 178–185.
Hjermind LE, Vissing J, Asmus F et al: No muscle involvement in myoclonus-dystonia caused by epsilon-sarcoglycan gene mutations. Eur J Neurol 2008; 15: 525–529.
Tsubata S, Bowles KR, Vatta M et al: Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest 2000; 106: 655–662.
Richardson P, McKenna W, Bristow M et al: Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996; 93: 841–842.
Felker GM, Thompson RE, Hare JM et al: Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med 2000; 342: 1077–1084.
Grunig E, Tasman JA, Kucherer H, Franz W, Kubler W, Katus HA : Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol 1998; 31: 186–194.
Mestroni L, Rocco C, Gregori D et al: Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J Am Coll Cardiol 1999; 34: 181–190.
Michels VV, Moll PP, Miller FA et al: The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 1992; 326: 77–82.
Franz WM, Muller OJ, Katus HA : Cardiomyopathies: from genetics to the prospect of treatment. Lancet 2001; 358: 1627–1637.
Osterziel KJ, Perrot A : Dilated cardiomyopathy: more genes means more phenotypes. Eur Heart J 2005; 26: 751–754.
Karkkainen S, Miettinen R, Tuomainen P et al: A novel mutation, Arg71Thr, in the delta-sarcoglycan gene is associated with dilated cardiomyopathy. J Mol Med 2003; 81: 795–800.
Sylvius N, Duboscq-Bidot L, Bouchier C et al: Mutational analysis of the beta- and delta-sarcoglycan genes in a large number of patients with familial and sporadic dilated cardiomyopathy. Am J Med Genet A 2003; 120 (Part A): 8–12.
Nigro V, Okazaki Y, Belsito A et al: Identification of the Syrian hamster cardiomyopathy gene. Hum Mol Genet 1997; 6: 601–607.
Coral-Vazquez R, Cohn RD, Moore SA et al: Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell 1999; 98: 465–474.
Dincer P, Bonnemann CG, Erdir Aker O et al: A homozygous nonsense mutation in δ-sarcoglycan exon 3 in a case of LGMD2F. Neuromuscul Disord 2000; 10: 247–250.
Gouveia TL, Kossugue PM, Paim JF et al: A new evidence for the maintenance of the sarcoglycan complex in muscle sarcolemma in spite of the primary absence of δ-SG protein. J Mol Med 2007; 85: 415–420.
Shiga K, Yoshioka H, Masumiya T, Kimura I, Takeda S, Imamura M : Zeta-sarcoglycan is a functional homologue of gamma-sarcoglycan in the formation of the sarcoglycan complex. Exp Cell Res 2006; 312: 2083–2092.
Heydemann A, Demonbreun A, Hadhazy M, Earley JU, McNally EM : Nuclear sequestration of delta-sarcoglycan disrupts the nuclear localization of lamin A/C and emerin in cardiomyocytes. Hum Mol Genet 2007; 16: 355–363.
Zhu X, Hadhazy M, Groh ME, Wheeler MT, Wollmann R, McNally EM : Overexpression of gamma-sarcoglycan induces severe muscular dystrophy. Implications for the regulation of Sarcoglycan assembly. J Biol Chem 2001; 276: 21785–21790.
Acknowledgements
We thank Professor Hanns Lochmüller, Dr Guy A MacGowan and Rachel Thompson for their thoughtful comments and their help in improving this article. This study was funded by Heart Research UK, Special Trustees of the Newcastle University Hospitals, the Muscular Dystrophy Campaign (MDC) and the German Ministry of Education and Research (BMBF, Bonn, Germany). Diagnostic facilities at the Newcastle Muscle Centre are supported by the National Commissioning Group (NCG) for rare neuromuscular disorders. RB and VS are members of the German Muscular Dystrophy Network (MD-NET 01GM0601) funded by the BMBF; MD-NET is a partner of TREAT-NMD (EC, 6th FP, proposal # 036825). KB and VS are members of MRC Centre for Neuromuscular Diseases.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of Interest
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Bauer, R., Hudson, J., Müller, H. et al. Does δ-sarcoglycan-associated autosomal-dominant cardiomyopathy exist?. Eur J Hum Genet 17, 1148–1153 (2009). https://doi.org/10.1038/ejhg.2009.17
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.17
Keywords
This article is cited by
-
S151A δ-sarcoglycan mutation causes a mild phenotype of cardiomyopathy in mice
European Journal of Human Genetics (2014)
-
δ-Sarcoglycan-deficient muscular dystrophy: from discovery to therapeutic approaches
Skeletal Muscle (2011)
