
Abstract
On the basis of the Human Cytogenetic Database, a computerized catalog of the clinical phenotypes associated with cytogenetically detectable human chromosome aberrations, we collected from the literature 102 cases with chromosomal aberrations and split hand/foot malformation or absent fingers/toes. Statistical analysis revealed a highly significant association (P<0.001) between the malformation and the chromosomal bands 4q32–q35, 5q15, 6q16–q22 and 7q11.2–q22 (SHFM1). Considering these findings, we suggest additional SHFM loci on chromosome 4q, 6q and probably 5q. The regions 4q and 6q have already been discussed in the literature as additional SHFM loci. We now show further evidence. In the proposed regions, there are interesting candidate genes such as, on 4q: HAND2, FGF2, LEF1 and BMPR1B; on 5q: MSX2, FLT4, PTX1 and PDLIM7; and on 6q: SNX3, GJA1, HEY2 and Tbx18.
Similar content being viewed by others


Introduction
Split hand/foot malformation (SHFM) is a malformation characterized by the abnormal development of the central rays of the distal limbs. Severity and clinical variability (syndactyly, median clefts of the hands and feet, and aplasia or hypoplasia of the phalanges, metacarpals and metatarsals) are highly variable.1 Falliner2 proposed a classification system of cleft hand with five types. These types differ in the involvement of thumb and small finger: from radial cleft hand with thumb aplasia, median cleft hand without involvement of thumb and little finger, to ulnar cleft hand with little finger aplasia. The incidence of SHFM is about 1:18 000. Five SHFM gene loci have been established for SHFM1–5, and at least one more locus exists, SHFM6.3 In 2006, Gurnett et al4 suggested an additional locus in the chromosomal region 8q21.11–q22.3, and Naveed et al5 detected novel susceptibility loci for split hand/foot malformation with long-bone deficiency (SHFLD) on chromosomes 1q42.2–q43 and 6q14.1. Other chromosomal regions and candidate genes have also been implicated.1, 6 Known SHFM loci refer to non-syndromic ectrodactyly; however, approximately 40% of SHFM patients have associated non-limb congenital anomalies.7 On the basis of the Human Cytogenetic Database (HCD), we present further evidence for at least two additional loci for SHFM on 4q32–q35 and 6q16–q22.
Materials and methods
Case selection
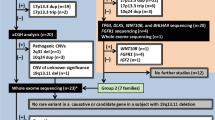
The HCD is an expertly curated computerized catalog of postnatally ascertained, cytogenetically detectable human chromosomal aberrations. The cases are collected from the literature since 1962. The first edition of HCD was published in 1994.8 A recent version of the HCD contains more than 9200 published cases with over 2800 different aberrations (unpublished data). The ‘International System for Human Cytogenetic Nomenclature’, a 400-band nomenclature,9 was used to describe the deletions and duplications. Deletions and duplications involving subbands were scored as including whole bands, and breakpoint bands were scored as deleted/duplicated. This gave a total of 7660 deleted and 10 183 duplicated bands at the end of 2005 (unpublished data). Our selection for studying SHFM was determined by the search mode of HCD (identical to London Dysmorphology Database). We not only collected cases with the description of split hand, split foot or ectrodactyly, but also we included the feature absent finger or toe (absent phalanges of one or more of the rays II–V) in our search. For the description of the selected cases, we used the classification system of Falliner.2 Cytogenetic and clinical data on individuals with non-mosaic simple deletions/duplications (those involving a single contiguous region of autosomal DNA) were extracted. Complex rearrangements were excluded from further analysis; this was done to avoid the known interactive effects in more complex rearrangements.10
Statistical analysis
Statistical analysis was carried out with SPSS Version 13.11 For the malformation studied, the observed number of deletions/duplications of a particular band was compared with the expected number calculated from the band distribution of all band deletions and duplications, respectively.12, 13, 14 We used crosstabs and calculated the P-values with Fisher's exact test (two-sided) to evaluate an association between deletion/duplication in a particular band and the presence of a malformation. Chromosomal bands found to be significantly associated were subdivided into two groups according to the P-values: P<0.01 (significant association) and P<0.001 (highly significant association). The candidate gene approach was used to identify genes, hemizygosity of which can result in SHFM. The list of genes localized on the bands found to be significantly associated with SHFM that were not previously reported was downloaded from Entrez Genome View (http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi; access October 2006). Candidates were selected according to known biological function, pattern of tissue expression and similar malformations in knockout organisms.
Results
A total of 35 cases (group S) with split hand, split foot or ectrodactyly were described in the HCD. In a total of 67 cases (group A), absent finger or toes were present without the mention of split hand/foot or ectrodactyly in the same individual. Thirty-seven cases (7 in the first and 30 in the second group) had to be excluded (complex rearrangements or mosaics).
Chromosomal aberrations involving 34 different bands in group S (Table 1), 81 different bands in group A (Table 2) and 94 different bands in the two groups together could be further analyzed. In group S, 73 bands were deleted and 12 duplicated, whereas in group A, the numbers of bands deleted and duplicated were 121 and 33, respectively.
By statistical analysis for group S, a highly significant association (P<0.001) was found for the bands 6q16–q22 and 7q11.1–q31. In group A, a significant association was present for the bands 13q21–q32 and a highly significant association for the bands 4q31.3–q34. Taking the two groups together, a significant association was found on 2q31–q32 and a highly significant association on 4q32–q35, 5q15, 6q16–q22 and 7q11.2–q22 (Table 3).
Discussion
The patients described in HCD most often present with multiple congenital anomalies based on a chromosomal aberration involving several different genes. We assume the concurrence of a certain malformation with a specific chromosomal aberration in most of the cases not being coincidental. Three studies with data from HCD have already been published in the last years, suggesting causative gene loci for different malformations.12, 13, 14 Interestingly, all the aberrations listed in the HCD with split hand, split foot or ectrodactyly concern known SHFM loci, except the cases on 4p, 4q, 5q, 6q, 12p and Trp(14)(pter->q12) (Table 1). The entire data from the HCD increased in our study the significance of this observation with additional numbers and by statistical means. In addition, the significant association with the known SHFM loci 7q21 and 2q31 confirms the validity of our study and we conclude that the other highly significantly associated regions on 5q15, 4q32–q35 and 6q16–q22 are also valuable candidate regions for further SHFM loci.
Some of the limitations of the statistical approach used in this study have already been discussed elsewhere.12, 13, 14 The statistical analysis assumes that each chromosome band can be treated independently. However, breakpoints involved in rearrangements are not randomly distributed and the role of predisposing low-copy repeats contributing to this disparity has been widely discussed.15 The effect of such ‘clustering’ of involved bands is to reduce the resolution of chromosomal maps, rather than to identify false loci. Another limitation is that any disease-causing gene locus close or even overlapping with a haplolethal region will not be detected with the strategy chosen. The same applies for locations of recessive genes: this method is not expected to reliably identify them. The aberrations collected in the HCD were published between 1962 and 2005. Some cytogenetic descriptions may be incomplete and the breakpoints described imprecise, as data from the ECARUCA project have shown.16 Owing to the fact that the proposed candidate regions refer to several independently described different case reports, we are still quite confident about the chromosomal regions. Future researchers have to consider that the breakpoints may be imprecise and, in the search for candidate genes, the neighboring segments have to be considered as well. This confirms once more the fact that in future studies of chromosomal aberrations, high-resolution molecular karyotyping, for example array comparative genomic hybridization, should be performed to refine the boundaries.
Comparing the collection of the HCD used in this study with the personal database of I W Lurie (unpublished data), only few cases of ectrodactyly and absent fingers were missing in our analyses. The missing cases involved aberrations of chromosome 2q,17, 18 7q19, 20 and 12p.21 As most of these six cases refer to known SHFM loci, they would not have changed our statistical conclusions.
Interestingly, of the 239 affected bands studied, 4/5 were deleted and only 1/5 duplicated. We conclude that, at least in our study population, the SHFM genes mainly act in association with haploinsufficiency. Embryopathy associated with segmental hypoploidy may be due to haploinsufficiency for a series of developmentally important genes.13 Owing to the lack of sufficient resolution in G-banding observations, many ‘duplications’ may be more complex rearrangements including deletions of candidate regions.
6q
The chromosomal region around band 6q21 was already discussed by different authors1, 22, 23, 24 as a candidate locus for SHFM, and Naveed et al5 recently presented the band 6q14.1 as a susceptibility locus for SHFLD. In our study, the chromosomal bands 6q16–q22 are highly significantly associated with SHFM. The malformations described in our affected cases involved primarily both upper limbs; the thumbs were always present, and the fifth finger was partly affected. In conclusion, the malformations could in general be regarded as medio-ulnar clefts. There are about 230 genes known in the region 6q16–q22 with interesting candidate genes such as SNX3, 6q21 (microcephaly, microphthalmia, ectrodactyly and prognathism1, 25), GJA1, 6q22.31 (oculodento digital dysplasia26), and HEY2, 6q22.31 (basic helix-loop-helix transcription factors control cell-fate decisions such as segmentation, neurogenesis and myogenesis in vertebrates27). In addition, there is one T-box gene, Tbx 18, on the long arm of chromosome 6 (6q14.3). The T-box family of transcriptional factors has major roles in embryogenesis, including the development of the limbs.28 Considering the possibility of imprecise breakpoint determination, we presume that the significant bands in our study and the earlier reports refer to the same SHFM locus on 6q.
4q
Lurie29 suggested an ectrodactyly gene on 4q33. We now have further evidence for a locus on 4q. In our study, deletions on band 4q32–q35 were highly significantly associated with absent fingers or toes, and at a closer look at the described cases and the involved rays, there was involvement of the median and ulnar rays (with the exception of one case, in which an absent thumb is described). These findings, which are mainly confined to the upper limbs, together with the mentioned syndactyly, fit very well to the definition of SHFM in general and medio-ulnar or ulnar cleft hands in particular. Interestingly, in most of the cases, only one limb, mainly the left upper limb, is affected.
In our study, the region 4q was not significantly associated with SHFM regarding the group S only. However, we consider 4q32–q35 as a valuable candidate region for SHFM, as the definition of SHFM includes an abnormal development of the central rays of the distal limbs. The clinical description of absent fingers II–V falls into the highly variable phenotype of SHFM and therefore we consider the data relevant taking the two groups A and S together. Relating to this observation, penetrance may be lower and/or clinical variability higher in any putative SHFM gene on 4q compared with 6q and 7q. Regarding our data, the relevance of 4q in SHFM seems to be at least comparable with the locus SHFM5 on 2q31.
There are about 180 genes known in the region, one of which is, for example, HAND2, 4q34.1 (expressed in developing vasculature and its derivative).30 In close proximity, there are other possible candidate genes, such as FGF2 (4q26), LEF1 (4q25), BMPR1B (4q22.3, brachydactyly type A) and potentially MED28 (4p15.32).
5q
In the HCD, there were only two duplications on the long arm of chromosome 5 in association with split hand/foot and absent toe registered. Considering the band 5q15, 2 of the 10 cases, with duplications described in the HCD, were affected, resulting in the statistically highly significant association. This significant association with SHFM in our study may be by chance. Nevertheless, there are interesting candidate genes near this region: MSX2 (5q35.2),31 FLT4 (5q35.3), PTX1 (5q31.1) and PDLIM7 (5q35.3).
13q
In our study, deletions at the chromosomal region 13q21–q31 were significantly associated with absent fingers II–V or toes II–V, whereas no patient with a chromosomal aberration on 13q and split hand or foot was described. The malformations of our affected cases are difficult to be classified according to Falliner;2 they seem to be symmetrical and likewise involve upper and lower limbs. As candidate genes for absent finger/toe within this region, we propose, for example, FGF9 (13q12.11, expressed in limb buds) and CDX2 (13q12.2, controlling embryonic axial elongation and anterior/posterior patterning).
Further molecular genetic studies are needed to examine whether there are SHFM families with linkage either to 4q, 5q or 6q. It also remains to be demonstrated that mutations involving the proposed candidate genes may cause split hand, split foot and/or absent phalanges of the rays II–V. Today we do not know the function and expression of all human genes and we have only limited data about knockout organisms, there may be more candidate genes in the regions than we mentioned.
In summary, we have further evidence for at least two additional loci for SHFM on chromosomes 4q32–q35 and 6q16–q22.
References
Duijf PHG, Van Bokhoven H, Brunner HG : Pathogenesis of split-hand/split-foot malformation. Hum Mol Genet 2003; 12: R51–R60.
Falliner A : Die Spalthand. Vorschlag einer Klassifikation auf der Basis von 279 Spalthänden. Handchir Mikrochir Plast Chir 2004; 36: 47–54.
Basel D, Kilpatrick MW, Tsipouras P : The expanding panorama of split hand foot malformation. Am J Med Genet 2006; 140A: 1359–1365.
Gurnett CA, Dobbs MB, Nordsieck EJ et al: Evidence for an additional locus for split hand/foot malformation in chromosome region 8q21.11-q22.3. Am J Med Genet 2006; 140A: 1744–1748.
Naveed M, Nath AK, Gaines M et al: Genomewide linkage scan for split-hand/foot malformation with long-bone deficiency in a large arab family identifies two novel susceptibility loci on chromosomes 1q42.2–q43 and 6q14.1. Am J Hum Genet 2007; 80: 105–111.
Elliott AM, Evans JA, Chudley AE : Split hand/foot malformation (SHFM). Clin Genet 2005; 68: 501–505.
Evans JA, Vitez M, Czeizel A : Congenital abnormalities associated with limb deficiency defects: a population study based on cases from the Hungarian Congenital Malformation Registry (1975–1984). Am J Med Genet 1994; 49: 52–66.
Schinzel A : Human cytogenetics database. Oxford Medical Databases Series. Oxford: Oxford University Press, 1994.
Mittelman F (ed): ISCN: an international system for human cytogenetic nomenclature. Basel: Karger, 1995.
Lurie IW : Autosomal imbalance syndromes: genetic interactions and the origins of congenital malformations in aneuploidy syndromes. Am J Med Genet 1993; 47: 410–416.
SPSS. Version 13, Copyright © 2000-2006, SPSS Schweiz AG.
Brewer C, Holloway S, Zawalnyski P, Schinzel A, FitzPatrick D : A chromosomal deletion map of human malformations. Am J Hum Genet 1998; 63: 1153–1159.
Brewer C, Holloway S, Zawalnyski P, Schinzel A, FitzPatrick D : A chromosomal duplication map of malformations: regions of suspected haplo- and triplolethality – and tolerance of segmental aneuploidy – in humans. Am J Hum Genet 1999; 64: 1702–1708.
Tyshchenko N, Lurie I, Schinzel A : Chromosomal map of human malformations. Hum Genet 2008; 124: 73–80.
Shaw CJ, Lupski JR : Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet 2004; 13 (Spec no 1): R57–R64.
Schinzel A : Review of chromosome aberration breakpoints in the context of ECARUCA (European cytogeneticists association register of unbalanced chromosome aberrations) project; 11th International congress of human genetics, Brisbane 2006: free paper abstract 0322, page 107.
Lurie IW, Gurevich DB, Autko NI : Del (2)(q31q33) syndrome; Proc 5th Meeting of the Belorussian Genetic Soc, Gorki 1986, 74 (Russian).
Del Campo M, Jones MC, Veraksa AN et al: Monodactylous limbs and abnormal genitalia are associated with hemizygosity for the human 2q31 region that includes the HOXD cluster. Am J Hum Genet 1999; 65: 104–110.
Scherer SW, Poorkaj P, Massa H et al: Physical mapping of the split hand/foot locus on chromosome 7 and implication in syndromic ectrodactyly. Hum Mol Genet 1994; 3: 1345–1354.
Fukushima K, Nagai K, Tsukada H et al: Deletion mapping of split hand/split foot malformation with hearing impairment: a case report. Int J Pediatr Otorhinolaryngol 2003; 67: 1127–1132.
Thomson LL, Stein CK, Shrimpton AE, Willey AM, Hoo JJ : A rare 12p distal interstitial deletion with radial ray anomaly of the hands and ectrodactyly of the feet: cytogenetic and molecular delineation. Am J Hum Genet 1996; 59 (Suppl): abstract 346.
Braverman N, Kline A, Pyeritz RE : Interstitial deletion of 6q associated with ectrodactyly. Am J Hum Genet 1993; 53 (Suppl): abstract 410.
Gurrieri F, Cammarata M, Avarello RM et al: Ulnar ray defect in an infant with a 6q21;7q31.2 translocation: further evidence of a limb defect gene in 6q21. Am J Med Genet 1995; 55: 315–318.
Correa-Cerro L, Garcia-Cruz D, Fiaz-Castanos L, Figuera LE, Sanchez-Corona J : Interstitial deletion 6q16.2q22.2 in a child with ectrodactyly. Ann Genet (Paris) 1996; 39: 105–109.
Vervoort VS, Viljoen D, Smart R et al: Sorting nexin 3 (SNX3) is disrupted in a patient with a translocation t(6;13)(q21;q12) and microcephaly, microphthalmia, ectrodactyly, prognathism (MMEP) phenotype. J Med Genet 2002; 39: 893–899.
Paznekas WA, Boyadjiev SA, Shapiro RE et al: Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet 2003; 72: 408–418.
Steidl C, Leimeister C, Klampt B et al: Characterization of the human mouse HEY1, Hey2, and HeyL genes: cloning, mapping, and mutation screening of a new bHLH gene family. Genomics 2000; 66: 195–203.
King M, Arnold JS, Shanske A, Morrow BE : T-genes and limb bud development. Am J Med Genet 2006; 140A: 1407–1413.
Lurie IW : Further study of genetic interactions: loss of short arm material in patients with ring chromosome 4 changes developmental pattern of del(4)(q33). Am J Med Genet 1995; 56: 308–311.
Yamagishi H, Olson EN, Srivastava D : The basic helix-loop-helix transcription factor, dHAND, is required for vascular development. J Clin Invest 2000; 105: 261–270.
Jabs EW, Müller U, Li X et al: A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell 1993; 75: 443–450.
Acknowledgements
We are grateful to Dr David FitzPatrick MD FRCP for critically reviewing the paper and enriching it with his comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Niedrist, D., Lurie, I. & Schinzel, A. 4q32–q35 and 6q16–q22 are valuable candidate regions for split hand/foot malformation. Eur J Hum Genet 17, 1086–1091 (2009). https://doi.org/10.1038/ejhg.2009.11
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.11
Keywords
This article is cited by
-
Novel and functional variants within the TBX18 gene promoter in ventricular septal defects
Molecular and Cellular Biochemistry (2013)
