
Abstract
In this report, we present three families in which we identified asymptomatic carriers of a homozygous absence of the SMN1 gene. In the first family, the bialleleic deletion was found in three of four siblings: two affected brothers (SMA type 3a and 3b) and a 25-years-old asymptomatic sister. All of them have four SMN2 copies. In the second family, four of six siblings are affected (three suffer from SMA2 and one from SMA3a), each with three SMN2 copies. The clinically asymptomatic 47-year-old father has the biallelic deletion and four SMN2 copies. In the third family, the biallelic SMN1 absence was found in a girl affected with SMA1 and in her healthy 53-years-old father who had five SMN2 copies. Our findings as well as those of other authors show that an increased number of SMN2 copies in healthy carriers of the biallelic SMN1 deletion is an important SMA phenotype modifier, but probably not the only one.
Similar content being viewed by others


Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder caused by loss of lower motor neurons.1 The estimated incidence of SMA is 1 per 7000–10 000 live births, corresponding to the prevalence of a mutated gene in 1 per 42–50 persons. Recent studies suggest a higher prevalence of the carrier state amounting to 1 per 34 persons.2, 3 The disease is characterized by significant phenotype variability. According to the International SMA Consortium, three clinical forms are distinguished depending on the age at onset and motor development milestones, designated as SMA1 (onset occurs within 6 months after birth and affected children are never able to sit unaided), SMA2 (onset occurs within 18 months and children are able to sit but they never walk) and SMA3 (weakness develops in patients who were previously able to walk unassisted).4 Type 3 SMA is further subdivided into two subgroups: SMA3a (onset below 3 years of age) and SMA3b (onset at the age ≥3 years). This distinction is related to the observation that children presenting with symptoms below 3 years of age lose ability to walk sooner than the children with symptoms over 3 years of age (about 30% in 3a versus 3% in 3b in the first 10 years after the onset of symptoms).5 Cases presenting with the first symptoms of the disease at the age of 20–30 years are classified as SMA4, or proximal adult type SMA.6, 7
All these forms of SMA result from low levels of the survival motor neuron (SMN) protein, which is insufficient to protect motor neurons.8 The SMN protein plays a major role in the metabolism of RNA.9 In addition, it probably has a specific function in the nervous system related to the neuromuscular junction maturation.10, 11 The SMN protein is coded by two genes, SMN1 and SMN2. In SMA patients, the SMN1 gene is mutated (a biallelic deletion occurs in more than 95% of patients) and the SMN protein is produced in reduced amounts by the SMN2 gene.12 The SMN2 gene may have several copies. In approximately 80% of the general population, one or two copies of the SMN2 gene are found.13, 14, 15 In SMA patients, the diversity in number of the SMN2 copies is much larger, ranging from 1 to 6 copies. The higher the number of SMN2 copies, the more benign is the SMA phenotype. Patients with the severe form of the disease usually have 1–2 copies, while patients with the intermediate form have 2–3 copies, and the patients with the mild or adult form have 3–4 or even 5–6 copies.3, 16 Thus the number of the SMN2 copies is undoubtedly the major SMA phenotype modifier. Some reports suggest that the presence of five SMN2 copies compensates the loss of both SMN1 gene alleles and may be responsible for the exceptionally rare asymptomatic biallelic deletion of the SMN1 gene.17 However fewer than five copies of SMN2 found in some asymptomatic carriers of a biallelic deletion of the exon 7 of the SMN1 gene suggest that other factors also play a role in the determination of the SMA phenotype.18
In this report, we present three asymptomatic carriers of biallelic absence of the SMN1 gene detected in three families affected with SMA. Two subjects had four SMN2 copies, which is the average number seen in the mild form of SMA, and one subject had five SMN2 copies.
Patients and methods
Molecular testing for a biallelic deletion in the SMN1 gene was performed in 490 clinically healthy members of families affected with SMA, including 386 parents (213 mothers and 173 fathers), 63 siblings (29 brothers and 34 sisters), 9 children (3 sons and 6 daughters) and 32 cousins, and also in 300 healthy controls (persons with a negative family history of neuromuscular disorders).
Methods
Genomic DNA was collected from peripheral blood of all family members. DNA was isolated from blood samples by the salting-out procedure.19
Homozygous deletion of exons 7 and 8 of SMN1 was detected using the restriction enzyme analysis, as described elsewhere.20
For SMN1 and SMN2 dosage analysis, the method described by Anhuf et al was employed with modification.14, 21, 22 Photometric measuring of DNA was performed on Gene Quant II (Pharmacia) spectrophotometer. DNAs were diluted to a concentration of 10 ng/μl. The same DNA samples of three normal individuals were used as a standard in all amplification reactions for SMN1 and SMN2. We used MGB probes to distinguish between SMN1 and SMN2. The MGB probes were specific for the SMN1 and the SMN2 at position 6 in exon 7. The SMN1 probe contained a VIC reporter dye connected to the 5′ end; the SMN2 probe contained a FAM reporter dye. Primers were identical for SMN1 and SMN2. As a reference gene (one copy, external control), we used an RN-ase P gene (TaqMan RN-ase P Control Reagents System Kit, Applied Biosystems). The RQ-PCR was performed on 96-well MicroAmp optical plates in a total volume of 25 μl that contained 50 ng of DNA, 300 nM of each primer, 100 nM of SMN1 and SMN2 probes and 12.5 μl of TaqMan Universal PCR Master Mix (Applied Biosystem). PCR was performed in the 7500 ABI Prism Sequence Detection System producing typical TaqMan reaction: 2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Every sample was analyzed for SMN1 and SMN2 genes and independently for the RN-ase P gene in triplets. Each reaction was duplicated and mean results were calculated. Date evaluation was carried out using Relative Quantification (ddCt) Study 7500 System SDS Software.
EMG was performed in six patients, in four of them it was analyzed quantitatively. EMG activity was estimated at rest, and at maximal voluntary contraction, at least 20 motor unit potentials (MUP) were collected in each muscle for quantitative analysis. Two patients were examined with Keypoint Medtronic and two with Counterpoint Medtronic systems. Collected data were compared with our laboratory's reference values. In the upper extremity the biceps brachii muscle was tested in three subjects, and the deltoid in one (family 2, I.1); in the lower extremity the rectus femoris muscle was tested.
The SMN protein levels in fibroblast cells were determined in eight members of families 1 and 2 and in seven healthy volunteers with no family history of the disease. The western blot analysis was performed with monoclonal anti-SMN antibodies (BD Biosciences; diluted 1:1000), and monoclonal anti-β-tubulin antibodies (Sigma-Aldrich Inc.; diluted 1:3000), as described previously.8 At least two independent cell lysates from cultured fibroblasts were analyzed, each in duplicate. The mean SMN level for the control group was assigned as 100%, and the final results for tested individuals were expressed as the mean percentage values. The mean value for the affected and unaffected patients was estimated and statistical analysis (Student's t-test) was performed.
Results
A biallelic deletion in the SMN1 gene was found in three clinically healthy members of families affected with SMA. We did not find a biallelic deletion in the SMN1 gene in any of 300 subjects from the control group.
Detailed characteristics of asymptomatic patients with biallelic deletion and their families is presented below.
Family 1 (no. 528)
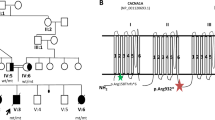
There are four siblings in family 1 (Figure 1a; Table 1). Two brothers (II4 and II5) are affected with SMA 3b and 3a, respectively. The diagnosis was confirmed by molecular testing (del. exon 7 and 8 of the SMN1 gene). Both showed neurogenic changes in upper and lower extremity muscles. Two other siblings are clinically healthy, they did not show any symptoms on neurological examination. EMG revealed chronic neurogenic changes in the lower extremity muscles in both of them (sister II-2: m. biceps: N; m. rectus femoris: amp. 2995; area 6542; 63% of outliners; brother II-3: m. biceps: N; m. vastus: 1228/1856; 18% of outliners). Molecular testing showed the biallelic deletion in a healthy sister (II-2) and a heterozygous deletion carrier state in a healthy brother (II-3). Both parents are deletion carriers (one SMN1 copy). A fifth child (II-1) born at 7 months of gestation died soon after birth.

Pedigrees of spinal muscular atrophy (SMA) families with biallelic absence of the SMN1 gene in unaffected subjects. (a) family 1, (b) family 2, (c) family 3; circles represent females, squares represent males, black symbols represent biallelic SMN1 absence, dots represent heterozygous SMA carriers, arrows represent asymptomatic carriers of biallelic SMN1 absence.
Family 2 (no. 899)
There are six siblings in this family (Figure 1b; Table 1), including three affected with SMA2, the youngest boy affected with SMA3a and two healthy ones. The parents are clinically healthy. Prenatal testing was carried out twice (both results were abnormal). Molecular testing showed a biallelic deletion of exons 7 and 8 of the SMN1 in the four affected siblings and in the clinically healthy father. EMG performed in the asymptomatic father (II–1) revealed chronic neurogenic changes in the lower extremity muscles (m. deltoid: N; m. rectus femoris amp.1680; area 3341). The mother and the healthy siblings are heterozygous deletion carriers.
Family 3 (no. 676)
In this family (Figure 1c; Table 1), a biallelic SMN1 gene deletion was found in a child with an acute form of SMA and in her asymptomatic father. The family was clinically examined only once and currently they are lost to follow-up and the affected child has died.
The SMN protein analysis showed no statistically significant differences between the mean value for the affected (84.0±9.5) and unaffected patients (94.5±12.9) (P=0.199) from SMA families.
Discussion
An asymptomatic biallelic deletion in the SMN1 gene is exceptionally rare. We found this deletion only in three cases among 490 clinically healthy relatives of SMA patients (including 437 obligatory deletion carriers, that is parents and offspring as well as siblings at a 2/3 chance of being heterozygous carriers) but in none of control group. Prior et al found the biallelic deletion in two cases among 408 deletion carriers.17 In a group of 214 parents and 66 siblings, Wang et al found a definite biallelic deletion in two clinically healthy subjects.23 Thus, the prevalence of an asymptomatic biallelic deletion carrier state may be estimated at approximately 0.5–0.7% of first-degree relatives of SMA patients. This is a low rate but highlights the need for genetic testing in members of families affected with SMA and bears importance to genetic counseling, since the presence of such a variant in one of the parents means that the risk of having an affected offspring is not 25 but 50% (as in the family 2 of this report).
To date, according to our knowledge, an asymptomatic biallelic deletion in the SMN1 gene has been reported in 23 subjects from 19 families.17, 18, 23, 24, 25, 26, 27, 28 In 13 families, siblings of asymptomatic carriers were affected, while in 5 families children of such carriers and in one family, a brother and a son of a carrier were affected. In this report, we present the following three cases of an asymptomatic biallelic deletion in the SMN1 gene.
Naturally we cannot exclude the possibility of a future occurrence of symptoms in these patients, particularly in view of their abnormal EMG results suggestive of an ongoing pathologic process. EMG data on the patients with asymptomatic biallelic deletion is scarce and electrophysiological methods employed differ between reports. Prior et al and Cobben et al described three patients with normal electrophysiological parameters.17, 24 Yet, one of the subjects was 6 months of age at the time of EMG, and one tested at 35 years of age had normal motor unit number estimate, but data on concentric needle EMG was not given. Mild neurogenic changes with increased MUP amplitude but no denervation were detected in 16- and 22-years old asymptomatic females.27, 28 The findings are not easy to interpret in view of the fact that mild neurogenic changes were also found in a healthy brother who is a heterozygous deletion carrier, with only one copy of SMN1 (subject II.3 in family 1). Mild neurogenic changes were previously reported in some parents of the children affected with SMA, who are nearly obligatory deletion carriers.29 The presence of neurogenic changes in unaffected heterozygous deletion carriers may suggest a subclinical defect resulting from SMN protein deficiency, detected with EMG. Yet, comparison of the severity of denervation/reinervation process between heterozygous deletion carriers is not possible from the published data, as EMG was reported in a qualitative way.24, 28 The lack of symptoms in subjects with the biallelic deletion in the SMN1 gene may be related to two factors, that is the increased number of SMN2 gene copies or the effect of other unknown phenotype modifiers. The presence of five SMN2 gene copies may explain the lack of clinical symptoms in the patient in family 3. Prior et al found five SMN2 gene copies in three asymptomatic carriers.17 However, the effect of the number of SMN2 gene copies is not absolute and the presence of five copies does not always compensate for the loss of the SMN1 gene. Wirth et al described three patients affected with a mild SMA form who had five or even six SMN2 copies.16 In addition, an increased number of SMN2 gene copies cannot explain the lack of clinical symptoms in cases in families 1 and 2. We found four copies of the SMN2 gene in these patients that is typical for SMA3. Molecular testing showed 2–3 SMN2 copies in asymptomatic carriers also in the families described by Helmken et al.18 In these families, the number of SMN2 gene copies was identical in affected subjects and their asymptomatic siblings. Thus, it seems that apart from SMN2 copies some other phenotype modifiers are also effective in SMA.
It is possible that gender may be one of the phenotype modifying factors. Twice lower incidence of SMA among girls above 8 years of age compared to age-matched boys was already reported long time ago.30 In contrast, biallelic deletion in unaffected subjects described earlier was more prevalent among females (14 females versus 9 males).17, 18, 23, 24, 25, 26, 27, 28 This is even more pronounced if only siblings (sharing identical genotype of SMA locus) are taken into account. Among 14 families, females were asymptomatic carriers in 9, while males in only 4 families, and one family included both a male and a female asymptomatic carrier (together 13 females versus 5 males). This gender proportion may be purely coincidental, but it may also suggest the presence of a gender-related protective factor.
Determination of the SMN protein level in family no. 528 showed nearly identical values in the affected brothers (83% and 94%) and their asymptomatic sister who was a carrier of the biallelic deletion (100%). In family no. 899, the SMN protein level ranged from 71 to 96% in the affected siblings and was 103% in the asymptomatic father. These differences between affected and unaffected biallelic deletion carriers were not statistically significant (P=0.199). It is in agreement with previous papers. The SMN protein levels have been shown to be not helpful to differentiate milder forms and controls, even in limphoblastoid cell lines.8, 31, 32 However Helmken et al showed that the protein level was higher in asymptomatic deletion carriers but only in lymphocytes, whereas fibroblasts showed identical protein expression in affected and unaffected siblings.18 In contrast, the total amount of the SMN transcript was identical in phenotypically discordant siblings. Thus, it seems that the SMN protein level may be tissue specific and phenotypic variability can be caused by changes at the translational level or through some completely different pathway.
The disease phenotype is undoubtedly influenced by the number of SMN2 copies. However, molecular DNA changes in the SMA region seem to be identical in both affected and asymptomatic carriers of the biallelic deletion, which was shown both in our cases and other similar subjects described earlier.17, 18, 23, 24, 25, 26, 27, 28 The varying phenotype may be related to some post-translational, epigenetic, perhaps tissue-specific changes affecting the SMN protein.
References
Dubowitz V : Disorders of Lower Motor Neurone: The Spinal Muscular Atrophies, In: Dubowitz V (ed): Muscle Disorders in Childhood. London: Saunders, 1995.
Cusin V, Clermont O, Gerard B, Chantereau D, Elion J : Prevalence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counselling. J Med Genet 2003; 40: e39.
Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B : Quantitative analysis of SMN1 and SMN2 based on real-time Light Cycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002; 70: 358–368.
Munsat T, Davies K : Report of international SMA consortium meeting. Neuromusc Disord 1992; 2: 423–428.
Zerres K, Rudnik-Schöneborn S, Forrest E et al: A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III): 569 patients. J Neurol Sci 1997; 146: 67–72.
Zerres K, Rudnik-Schöneborn S, Forkert R et al: Genetic bases of adult onset spinal muscular atrophy. Lancet 1995; 346: 1162.
Brahe Ch, Servidei S, Zappata S et al: Genetic homogeneity between childhood-onset and adult-onset autosomal recessive spinal muscular atrophy. Lancet 1995; 346: 741–742.
Lefebvre S, Burlet P, Liu Q et al: Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997; 16: 265–269.
Terns MP, Terns RM : Macromolecular complexes: SMN—the master assembler. Curr Biol 2001; 11: R862–R864.
Fan L, Simard LR : Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet 2002; 11: 1605–1614.
Cifuentez Diaz C, Nicole S, Velasco ME et al: Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet 2002; 11: 1439–1447.
Lefebvre S, Bürglen L, Reboullet S : Identification and characterization of spinal muscular atrophy determining gene. Cell 1995; 80: 155–165.
Gerard B, Ginet N, Matthijs G et al: Genotype determination at the survival motor neuron locus in normal population and SMA carriers using competitive PCR and primer extension. Hum Mut 2000; 16: 253–263.
Anhuf D, Eggermann T, Rudnik-Schöneborn S, Zerres K : Determination of SMN1 and SMN2 copy number using TaqMan technology. Hum Mut 2003; 22: 74–78.
Jędrzejowska M, Jurek M, Hausmanowa-Petrusewicz I : SMN1 and SMN2 copy numbers in Polish population. Neromusc Disord 2006; 16 (S1): S117.
Wirth B, Brichta L, Schrank B et al: Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet 2006; 119: 422–428.
Prior TW, Swoboda KJ, Scott HD, Hejmanowski AQ : Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet 2004; 130A: 307–310.
Helmken C, Hofmann Y, Schoehnen F et al: Evidence for modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet 2003; 114: 11–21.
Miller SA, Dykes DD, Poleski HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res 1988; 16: 1215.
Scheffer H, Cobben JM, Matthijs G, Wirth B : Best practice guidelines for molecular analysis in spinal muscular atrophy. Eur J Hum Genet 2001; 9: 484–491.
Jedrzejowska M, Wiszniewski W, Zimowski J et al: Application of a rapid non-invasive technique in the molecular diagnosis of spinal muscular atrophy (SMA). Neurol Neurochir Pol 2005; 39: 89–94.
Kim JY, Hwang JM, Ko HS et al: Mitochondrial DNA content is decreased in autosomal dominant optic atrophy. Neurology 2005; 64: 966–972.
Wang ChH, Xu J, Carter TA et al: Characterization of survival motor neuron (SMNT) gene deletions in asymptomatic carriers of spinal muscular atrophy. Hum Mol Genet 1996; 5: 359–365.
Cobben JM, Van der Steege G, Grootscholten P, De Visser M, Scheffer H, Buys CHCM : Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am J Hum Genet 1995; 57: 805–808.
Hahnen E, Forkert R, Marke Ch et al: Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum Mol Genet 1995; 4: 1927–1933.
Capon F, Levato C, Merlini L et al: Discordant clinical outcome in type III spinal muscular atrophy sibships showing the same deletion pattern. Neuromusc Disord 1996; 6: 261–264.
Bussaglia E, Tizzano EF, Illa I, Cervera C, Baiget M : Cramps and minimal EMG abnormalities as preclinical manifestation of spinal muscular atrophy patients with homozygous deletions of the SMN gene. Neurology 1997; 48: 1443–1445.
Cuscó I, Barceló MJ, Rojas-García R et al: SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J Neurol 2006; 253: 21–25.
Emery AEH, Anderson AR, Noronha MJ : Electromyographic studies in parents of children with spinal muscular atrophy. J Med Genet 1973; 10: 8–10.
Hausmanowa-Petrusewicz I, Zaremba J, Borkowska J, Szirkowiec W : Chronic proximal spinal muscular atrophy of childhood and adolescence: sex influence. J Med Genet 1984; 21: 447–450.
Patrizi AL, Tiziano F, Zappata S, Donati MA, Neri G, Brahe C : SMN protein analysis in fibroblast, amniocyte and CVS cultures from spinal muscular atrophy patients and its relevance for diagnosis. Eur J Hum Genet 1999; 7: 301–309.
Coovert DD, Le TT, McAndrew PE et al: The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 1997; 6: 1205–1214.
Acknowledgements
This study was supported by grants from the Polish State Scientific Committee (KBN) 2P05E no. 2P05E 007 27 (to MJ) and the Foundation to Support the Development of Polish Pharmacy and Medicine no. II/201/2003 (to IHP). Sequences of primers and probes for SMN1 and SMN2 genes were kindly donated by Francesco Danilo Tiziano (Policlinico Gemelli, Roma, Italy).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jędrzejowska, M., Borkowska, J., Zimowski, J. et al. Unaffected patients with a homozygous absence of the SMN1 gene. Eur J Hum Genet 16, 930–934 (2008). https://doi.org/10.1038/ejhg.2008.41
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.41
Keywords
This article is cited by
-
MLPA analysis for molecular diagnosis of spinal muscular atrophy and correlation of 5q13.2 genes with disease phenotype in Egyptian patients
Egyptian Journal of Medical Human Genetics (2022)
-
Observation of the natural course of type 3 spinal muscular atrophy: data from the polish registry of spinal muscular atrophy
Orphanet Journal of Rare Diseases (2021)
-
Perspectives in genetic counseling for spinal muscular atrophy in the new therapeutic era: early pre-symptomatic intervention and test in minors
European Journal of Human Genetics (2019)
-
Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype
Human Genetics (2019)
-
Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy – a literature review
Orphanet Journal of Rare Diseases (2017)
