
Abstract
Adenylosuccinate lyase deficiency is a rare autosomal disorder of de novo purine synthesis, which results in the accumulation of succinylpurines in body fluids. Patients with adenylosuccinate lyase deficiency show a variable combination of mental retardation, epilepsy and autistic features and are usually discovered during screens for unexplained encephalopathy using the Bratton–Marshall assay that reveals the excretion of the succinylaminoimidazolecarboxamide riboside (SAICAr). Here, we report on two sisters aged 11 and 12 years presented with global developmental delay, motor apraxia, severe speech deficits, seizures and behavioural features, which combined excessive laughter, a very happy disposition, hyperactivity, a short attention span, the mouthing of objects, tantrums and stereotyped movements that gave a behavioural profile mimicking Angelman syndrome. Both patients had an increased succinyladenosine/SAICAr ratio of 1.6, and exhibited a novel homozygous missense mutation (c.674T>C; p.Met225Thr) in the exon 6 of the ADSL gene. We suggest that these clinical features might be a new presentation of adenylosuccinate lyase deficiency. On the basis of this observation, although adenylosuccinate lyase deficiency is a rare disorder, this diagnosis should be considered in patients with mental retardation and a behavioural profile suggestive of Angelman syndrome.
Similar content being viewed by others
Introduction
Adenylosuccinate lyase (ADSL OMIM 103050) deficiency is a rare inborn error of purine metabolism leading to a broad spectrum of neurological disorders, ranging from fatal neonatal encephalopathy with hypokinesia to mild mental retardation with autistic features.1, 2, 3, 4, 5 Although clinical presentation is quite variable, most patients show at least marked mental retardation and hypotonia.6, 7, 8 In addition, most patients show autistic features, with repetitive behaviour, agitation, temper tantrums and autoagressivity.2, 9
ADSL catalyses two steps in the de novo purine pathway, specifically the conversion of succinylaminoimidazolecarboxamide ribonucleotide (SAICAR) into aminoimidazolecarboxamide ribonucleotide (AICAR), and the conversion of succinyladenosine monophosphate (S-AMP) to adenosine monophosphate (AMP). As a result of enzyme deficiency, the accumulating substrates, SAICAR and S-AMP, are dephosphorylated to SAICA riboside (SAICAr) and succinyladenosine (S-Ado), respectively and excreted into the plasma, urine and cerebral fluid (for review, see Spiegel et al9).
Patients with ADSL deficiency are discovered during screens of children with unexplained encephalopathy using the Bratton–Marschall assay for diazotizable amines.10 This assay reveals the excretion of SAICAr, which is the main criterion for the diagnosis. However, because of the possible false-positive results, the diagnosis should be confirmed by the identification of S-Ado and SAICAr in urine by high-performance liquid chromatography. Screens for mutations in the ADSL gene are also performed.
So far, 56 cases exhibiting more than 40 different disease-causing mutations have been reported, with a high prevalence of missense mutations leading to incomplete impairment of enzymatic activity.11 Correlations have been performed between the ADSL gene mutation or ADSL activity and the severity of the phenotype.9, 12, 13 More severe presentations tend to be associated with S-Ado/SAICAr ratio <2, whereas in milder clinical pictures, these ratios range from 2 to 4. This suggests that SAICAr is the offending compound, and that S-Ado could counteract its toxic effects.3 However, these results show a high level of variability and cannot always be exactly correlated with the clinical features.
Here, we report on two sisters with ADSL deficiency with severe mental retardation, common dysmorphic features and a specific behavioural pattern. An originality of our observations is the common behavioural features of these patients that could be suggestive of Angelman syndrome (AS).
Clinical reports
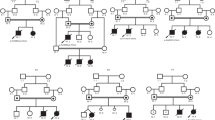
Both girls are sisters from consanguineous parents of Moroccan origin. The pedigree of the family is shown in Figures 1 and 2.

Pedigree of family.

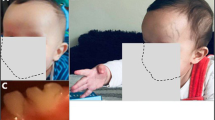
Facial dysmorphy in our cases. Note the wide mouth, wide-spaced teeth and prominent mandible in both sisters V-1 (a) and V-2 (b).
Case V-1 is a 11-year-old girl with severe mental retardation and disturbed behaviour. Spontaneous delivery was at term with birth weight (3300 g), length (49 cm) and head circumference (34 cm) in the normal range (50th percentile). The girl was described as being an unresponsive infant and delays of gross motor function and language were apparent during the first year of life. She was able to stand with support at the age of 3 years, started walking independently at the age of 4 years with gait ataxia. She achieved normal hand use (pincer grasp), and had a good eye-to-eye contact, but did not use any words until the age of 9 years.
At present, height (1.40 m) and weight (35 kg) are normal (50th percentile) with relative microcephaly (50 cm; 10th percentile). Dysmorphic features were noticed with a brachycephaly, divergent strabismus, wide mouth, wide-spaced teeth and a prominent mandible. Neurological examination disclosed severe axial hypotonia with hyperlaxity, combined with gait ataxia and brisk reflex without spasticity. Severe speech impairment with a minimal use of words was noticed that contrasted with a higher degree of receptive and non-verbal communication skills. Behavioural disturbances started during infancy and mainly consisted of an apparent happy demeanour with episodes of inappropriate laughter and hyperactivity, restlessness, easily excitable personality, mouthing objects and stereotypic hand movements consisting of the combination of hand flapping, mouthing and clapping. No sleep disorder was reported, nor self-aggressive, clastic and screaming episodes. Generalised tonic and clonic seizures started at the age of 9 years and were easily controlled with valproate. Repeated interictal EEG were normal. Brain magnetic resonance imaging (MRI) showed a moderate cerebral atrophy.
Case V-2, the proband's sister aged 12 years, exhibits a similar but milder phenotype. The neonatal period was normal and growth parameters were within the normal range (birth weight: 3500 g; length: 51 cm; head circumference: 35 cm). From the first months of life, severe hypotonia was observed with a poor eye-to eye contact and postural delay, but the patient never experienced regression. She learned to walk at the age of 3 years, but with a marked broad-based gait. Hand use was approximately normal. At present, growth parameters are within the normal range (weight 39 kg; height 1.50 m), although a secondary relative microcephaly (51 cm; 10th percentile) was also observed. Dysmorphic features were the same as her sister. Neurological examination showed hypotonia, gait ataxia and brisk reflex. Severe mental retardation was present with severe language impairment with better receptive skills. Behavioural disturbances started at the age of 3 years with the same combination of hyperactivity, an apparent happy demeanour with unprovoked laughter and stereotypic hand movements similar to her sister. This child, however, has never presented epileptic seizures.
As both sisters had the combination of severe mental retardation, absence of speech and behaviour that was reminiscent of AS, their karyotypes, methylation tests for AS and UBE3A sequencing were performed showing normal results.
Other members of this family were not evaluated. However, the family reported that the case V-3, the proband's brother, died at 10 years of age of strangulating intestinal occlusion, without any neuromotor impairment. In addition, case III-3 presented severe progressive encephalopathy with spastic quadriparesis and epilepsy that lead to death at 27 years and the case III-2, also presented moderate mental retardation with disturbed behaviour and a protected employee. The rest of the family was considered as normal. In particular, all these cases had a regular job and no psychiatric disorders.
Results
The modified Bratton–Marshall urinary test was positive suggesting ADSL deficiency. The diagnosis was confirmed by the measurement of urine S-Ado and SAICAr by HPLC. Urinary S-Ado and SAICAr concentration were 101 and 63 mmol mol−1 creatinine in case V-1 and 79 and 49 mmol mol−1 creatinine in case V-2, respectively. The S-Ado/SAICAr ratio was 1.6 for both patients.
Direct sequencing of the ADSL gene revealed in the two sisters the presence of a homozygous missense mutation, c.674T>C, located in exon 6 leading to the amino-acid substitution, p.M225T, at the protein level.
Discussion
To our knowledge, the original neurobehavioral presentation we report here in two sisters is the first such report in ADSL-deficient patients. Although the clinical presentation of ADSL deficiency is highly variable, previous reports have already underlined that half of the patients exhibited behavioural phenotypes that mainly consisted of autistic features, combined with mental retardation and hypotonia.1, 2, 3, 4, 6, 7 Both patients we report present mild dysmorphic features that were also previously mentioned in ADSL deficiency, although thought to be non-specific.14 The most severely affected sister (case V-1) also presented controlled generalised epilepsy that is also a classical hallmark in ADSL deficiency.8
The most striking feature of our observation is the behavioural phenotype that at first sight, is suggestive of AS.15, 16, 17 Indeed, the behavioural profile observed here, characterized in both sisters by hyperactivity, inappropriate laughter, a happy disposition and polymorphic hand stereotypies combined with severe mental retardation, profound speech impairment and ataxic gait clearly contrast with previous reports.15, 16, 17 By contrast, in the literature, half of the ADSL patients demonstrated autistic features with poor eye contact, hetero-aggressivity, repetitive behaviours, agitation and temper tantrums.2
Although the behaviour was reminiscent of AS, other consistent features of AS were not present. In particular, the characteristic EEG pattern of AS consisting of rhythmic triphasic waves of high amplitude was absent, as well as early onset seizures. In addition, sleep disturbances were absent. Finally, our patient did not demonstrate severe microcephaly and characteristic dysmorphic features that are observed in 20–80% patients according to Williams criteria.15, 16, 17
Analysis of behavioural features also referred to as ‘behavioural neurogenetics’ in neurodevelopmental disorders allows the delineation of distinctive behavioural phenotypes. Firstly, it permits to delineate profiles of social traits that can emerge as prominent syndromic features.18 Secondly, it can contribute to a better understanding of the biochemical bases of the disease. Finally, ‘behavioural neurogenetics,’ provides new insights into understanding how both genetic and metabolic factors contribute to neurodevelopmental pathways in learning and developmental disabilities.19
Considering ADSL deficiency, several metabolic alterations are believed to contribute to the neurobehavioural impairment. Of these, the most important factors could be the neurotoxic effects of accumulated succinylpurines.3 In both reported patients, a low S-Ado/SAICAr ratio of 1.6 was found, in agreement with the prediction of a severe phenotype. Other biochemical factors are the deficient synthesis of purine nucleotides and the impairment of the purine nucleotide cycle that contribute to the neurobehavioral profile in ADSL deficiency. As the S-Ado/SAICAr ratio was the same in both sisters, these additional factors could account for the relative disparity in their clinical presentations (for review see Spiegel et al9).
In conclusion, our observation confirms the wide phenotypic variability observed in ADSL deficiency, and highlights the importance of testing with an inexpensive urinary screening method, such as the modified Bratton–Marshall test in patients that show unexplained, profound as well as mild psychomotor retardation in neurological diseases with convulsions and/or hypotonia and in behavioural features of unknown origin.
References
Jaeken J, Van den Berghe G : An infantile autistic syndrome characterised by the presence of succinylpurines in body fluids. Lancet 1984; 2: 1058–1061.
Van den Berghe G, Vincent MF, Jaeken J : Inborn errors of the purine nucleotide cycle: adenylosuccinase deficiency. J Inherit Metab Dis 1997; 20: 193–202.
Jaeken J, Wadman SK, Duran M et al: Adenylosuccinase deficiency: an inborn error of purine nucleotide synthesis. Eur J Pediatr 1988; 148: 126–131.
van den Bergh FA, Bosschaart AN, Hageman G, Duran M, Tien Poll-The B : Adenylosuccinase deficiency with neonatal onset severe epileptic seizures and sudden death. Neuropediatrics 1998; 29: 51–53.
Mouchegh K, Zikanova M, Hoffmann GF et al: Lethal fetal and early neonatal presentation of adenylosuccinate lyase deficiency: observation of 6 patients in 4 families. J Pediatr 2007; 150: 57.e2–61.e2.
Maaswinkel-Mooij PD, Laan LA, Onkenhout W, Brouwer OF, Jaeken J, Poorthuis BJ : Adenylosuccinase deficiency presenting with epilepsy in early infancy. J Inherit Metab Dis 1997; 20: 606–607.
Sebesta I, Krijt J, Kmoch S, Hartmannova H, Wojda M, Zeman J : Adenylosuccinase deficiency: clinical and biochemical findings in 5 Czech patients. J Inherit Metab Dis 1997; 20: 343–344.
Nassogne M, Henrot B, Aubert G et al: Adenylosuccinase deficiency: an unusual cause of early onset epilepsy associated with acquired microcephaly. Brain Dev 2000; 22: 383–386.
Spiegel EK, Colman RF, Patterson D : Adenylosuccinate lyase deficiency. Mol Genet Metab 2006; 89: 19–31.
Laikind PK, Seegmiller JE, Gruber HE : Detection of 5′-phosphoribosyl-4-(N-succinylcarboxamide)-5-aminoimidazole in urine by use of the Bratton–Marshall reaction: identification of patients deficient in adenylosuccinate lyase activity. Anal Biochem 1986; 156: 81–90.
Van den Bergh F, Vincent MF, Jaeken J, Van den Berghe G : Residual adenylosuccinase activities in fibroblasts of adenylosuccinase-deficient children: parallel deficiency with adenylosuccinate and succinyl-AICAR in profoundly retarded patients and non-parallel deficiency in a mildly retarded girl. J Inherit Metab Dis 1993; 16: 415–424.
Marie S, Race V, Vincent MF, Van den Berghe G : Adenylosuccinate lyase deficiency: from the clinics to molecular biology. Adv Exp Med Biol 2000; 486: 79–82.
Race V, Marie S, Vincent MF, Van den Berghe G : Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum Mol Genet 2000; 9: 2159–2165.
Holder-Espinasse M, Marie S, Bourrouillou G et al: Towards a suggestive facial dysmorphism in adenylosuccinate lyase deficiency? J Med Genet 2002; 39: 440–442.
Williams CA, Beaudet AL, Clayton-Smith J et al: Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A 2006; 140: 413–418.
Barry RJ, Leitner RP, Clarke AR, Einfeld SL : Behavioral aspects of Angelman syndrome: a case control study. Am J Med Genet A 2005; 132: 8–12.
Williams CA, Lossie A, Driscoll D : Angelman syndrome: mimicking conditions and phenotypes. Am J Med Genet 2001; 101: 59–64.
Feinstein C, Singh S : Social phenotypes in neurogenetic syndromes. Child Adolesc Psychiatr Clin N Am 2007; 16: 631–647.
Baumgardner TL, Green KE, Reiss AL : A behavioral neurogenetics approach to developmental disabilities: gene-brain-behavior associations. Curr Opin Neurol 1994; 7: 172–178.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest
The authors state no conflicts of interest. The authors confirm that we have read the Journals position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Rights and permissions
About this article
Cite this article
Gitiaux, C., Ceballos-Picot, I., Marie, S. et al. Misleading behavioural phenotype with adenylosuccinate lyase deficiency. Eur J Hum Genet 17, 133–136 (2009). https://doi.org/10.1038/ejhg.2008.174
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.174
Keywords
This article is cited by
-
Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes
European Journal of Human Genetics (2019)
-
A mild form of adenylosuccinate lyase deficiency in absence of typical brain MRI features diagnosed by whole exome sequencing
Italian Journal of Pediatrics (2017)
-
Adenylosuccinate lyase deficiency
Journal of Inherited Metabolic Disease (2015)
-
Magnetic resonance imaging of the brain in adenylosuccinate lyase deficiency: a report of seven cases and a review of the literature
European Journal of Pediatrics (2012)
-
Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes
BMC Medical Genetics (2010)
