
Abstract
Congenital hearing loss affects approximately one child in 1000. About 10% of the deaf population have Usher syndrome (USH). In USH, hearing loss is complicated by retinal degeneration with onset in the first (USH1) or second (USH2) decade. In most populations, diagnostic testing is hampered by a multitude of mutations in nine genes. We have recently shown that in French Canadians from Quebec, USH1 largely results from a single USH1C founder mutation, c.216G>A (‘Acadian allele’). The genetic basis of USH2 in Canadians of French descent, however, has remained elusive. Here, we have investigated nine USH2 families from Quebec and New Brunswick (the former Acadia) by haplotype analyses of the USH2A locus and sequencing of the three known USH2 genes. Seven USH2A mutations were identified in eight patients. One of them, c.4338_4339delCT, accounts for 10 out of 18 disease alleles (55.6%). This mutation has previously been reported in an Acadian USH2 family, and it was found in homozygous state in the three Acadians of our sample. As in the case of c.216G>A (USH1C), a common haplotype is associated with c.4338_4339delCT. With a limited number of molecular tests, it will now be possible in these populations to estimate whether children with congenital hearing impairment of different degrees will develop retinal disease – with important clinical and therapeutic implications. USH2 is the second example that reveals a significant genetic overlap between Quebecois and Acadians: in contrast to current understanding, other genetic disorders present in both populations are likely based on common founder mutations as well.
Similar content being viewed by others


Introduction
Usher syndrome (USH) is an autosomal recessive condition characterized by sensorineural hearing loss, vestibular dysfunction, and visual impairment due to retinitis pigmentosa (RP). It is the leading cause of deafblindness, with a general prevalence of 2–6.2 in 100 000. Among the deaf population, the proportion of patients with USH may be as high as 10%.1 Three clinical subtypes are distinguished, with type 1 (USH1) representing the most severe subtype with profound congenital deafness, vestibular dysfunction, and early (prepubertal) onset of RP. Usher type 2 (USH2) is less severe and characterized by congenital, moderate to severe hearing impairment, normal vestibular function, and RP with onset in late adolescence (reviewed in Cohen et al1). Usher type 3 (USH3) presents with progressive, postlingual hearing loss, RP of variable onset and severity, and with or without vestibular impairment.
Three genes (USH2A, GPR98, and DFNB31) are currently known to harbor mutations in USH2, and most USH2 patients have mutations in the USH2A gene. The protein encoded by USH2A, usherin, is a transiently expressed component of interstereocilia ankle links in cochlear hair cells, suggesting an important role for their maturation.2 In mammalian photoreceptors, usherin localizes to a recess that is wrapped around the connecting cilium and may fulfill important structural and signaling roles.3 The c.2299delG mutation in USH2A is found in 16–77% of USH2A families (depending on the respective population).1 In addition, many, often private, USH2A mutations that are scattered throughout this large gene have been reported. Mutations in the other two known USH2 genes, DFNB31 (USH2D) and GPR98 (USH2C; the gene was formerly designated VLGR1b or MASS1), are rare.4, 5
The French Canadian population of Quebec (currently approximately 6 million people) descends from approximately 8500 French settlers who colonized the St Lawrence River valley between 1608 and 1759. For many Mendelian diseases in French Canadians, founder mutations have been identified.6 Mutations rare elsewhere are common in Quebec, and rare diseases elsewhere are common in the French-Canadian population, illustrating the unique genetic nature of this ‘island population’.6 We have recently shown that USH1 in Quebec is caused by a limited number of founder mutations, predominated by an USH1C mutation that is rare in the rest of the world.7 Here, we hypothesized that founder mutations in USH2 genes are also likely to exist in Canadians of French origin (from Quebec and New Brunswick), and we have elucidated the spectrum of mutations. We found a predominance of one truncating USH2A allele in both Quebecois and Acadians, which parallels the situation previously reported for USH1 and has important implications for future health-care strategies for hearing-impaired patients in these populations. One patient was found to carry two mutations in the USH2C gene, GPR98.
Methods
Patients
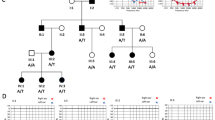
Nine Canadian families of French origin (seven from Quebec and two from New Brunswick, the former Acadia) with USH2 were identified through the McGill Ocular Genetics Laboratory, Montreal Children's Hospital Research Institute, McGill University Health Center, Montreal, Quebec, Canada (see Figure 1 for precise origin of each patient). The study was performed according to the Declaration of Helsinki and approved by the institutional review boards of both institutions involved (ethics committees of McGill University and the University Hospital of Cologne). Written informed consent was obtained from all participants. All patients met the diagnostic criteria for USH2. The healthy control individuals (all with a negative family history for USH) came from all regions of Quebec.

Map of Quebec and New Brunswick (the former Acadia). Numbers indicate patients investigated in this study. USH2A mutation alleles are encoded by (a–g). One patient was found to carry two mutations in the USH2C gene, GPR98 (genetic and clinical details of this patient will be described elsewhere).
Mutation and haplotype analysis
To detect founder mutations in USH2 patients from Quebec and New Brunswick, we first genotyped three polymorphic microsatellite markers, two flanking the USH2A locus (D1S474 and D1S490) and one within the gene (we have identified a novel polymorphic CA repeat in intron 44; primers available on request). In case of homozygosity for the alleles of the three marker, all coding exons (with the adjacent intronic regions) of the USH2A gene (71 coding exons) were sequenced. For patient MOGL 3065, where no mutation could be identified by this approach, the DFNB31 gene (USH2D; 12 coding exons) and GPR98 (USH2C; 90 coding exons) were analyzed by direct sequencing. To determine the carrier frequency of the USH2A deletion c.4338_4339delCT in the healthy population of Quebec, fluorescent dye-labeled primers were used for amplification of exon 3, and the PCR products from 207 healthy French-Canadian individuals were subsequently analyzed for their length on the ABI-377 DNA sequencer. Of which, 50 healthy control persons were genotyped for the novel truncating mutations identified in this study, whereas 157 controls were genotyped in case of the novel missense mutation p.T4809I. To elucidate the USH2A c.4338_4339delCT haplotype, we analyzed genotypes of 15 intragenic single nucleotide polymorphisms (SNPs; Table 1).
Results and discussion
Genotyping of three polymorphic microsatellite markers specific for the USH2A locus revealed homozygosity for the same haplotype in affected persons from four out of our nine pedigrees. Analysis of the coding region of the USH2A gene identified a homozygous truncating 2-bp deletion in exon 20, c.4338_4339delCT (p.C1447QfsX29), in all four of them. This mutation was also present in heterozygous state in two additional families, altogether accounting for 10 of 18 disease alleles (55.6%). Of note, it was present in homozygous state in all three of our Acadian USH2A pedigrees, which is in agreement with two previous reports that described this particular mutation in another Acadian USH2 family.8, 9 We identified no carriers of c.4338_4339delCT in 207 healthy control persons from Quebec.
We then identified six additional USH2A mutations in our cohort (Figure 2): c.612_613insG (p.R205EfsX11; exon 3), p.R626X (c.1876C>T, exon 11), c.2299delG (p.E767SfsX21; exon 13), c.7265_7269delTAACT (p.I2422RfsX38; exon 38), p.T4809I (c.14426C>T, exon 66), and p.R4935X (c.14803C>T, exon 68) were each found on one allele (6%). Besides the well-known c.2299delG mutation, both nonsense mutations have been described previously: p.R626X has been reported in Scandinavian and French patients as well as in American patients of Northern European ancestry.9, 10, 11 Baux et al11 have reported the p.R4935X mutation in one patient.

Cartoon of the usherin protein. Mutations identified in this study are depicted. Designations of domains are given above the scheme. IC, intracellular; PBM, PDZ-binding motif.
However, the two truncating mutations c.612_613insG and c.7265_7269delTAACT (not found in 50 French Canadian controls), as well as the missense mutation p.T4809I, are novel. The p.T4809I mutation was not found in 157 control individuals from Quebec, and it affects an evolutionarily highly conserved FN3 repeat. The p.T4809 residue is strongly conserved among vertebrates (such as human, chimpanzee, mouse lemur, northern treeshrew, domestic cat, dog, cattle, mouse, opossum, and chicken; (http://www.ensembl.org/Homo_sapiens/alignview?class=Homology;gene=ENSG00000042781). Usherin is involved in the composition of the ankle links, filamentous lateral links connecting hair cell stereocilia, thereby probably fulfilling a crucial role for hair cell maturation. FN3 repeats are present in proteins that experience mechanical stress in vivo, where they can help to maintain structural integrity.2 Although we consider p.T4809I as likely pathogenic (eventually interfering with the assumed usherin functions discussed above), we cannot exclude that it is a rare benign variant and that the second mutant allele is in a noncoding region of USH2A and escaped detection.
The six USH2 patients carrying the c.4338_4339delCT mutation all share a common ancestral haplotype (Table 1), which is in line with the concept of a founder mutation. The c.2299delG-associated haplotype in our patient MOGL 415 corresponds to the haplotype reported earlier in patients of geographically diverse origins (Dreyer et al12 found solely this haplotype to be associated with c.2299delG in 116 patients from 14 countries).
In summary, we found biallelic USH2A mutations in eight of our nine USH2 families of French origin (88.9%). One patient (MOGL 3065) was found to be compound heterozygous for two novel truncating mutations in the USH2C gene, GPR98. The detailed genotype and the clinical findings in this patient are described in detail elsewhere.
From a worldwide perspective, most patients with USH2 harbor mutations in USH2A,8, 13 a large gene (71 coding exons encoding a protein of 5202 amino acids) encoding a transmembrane protein that interacts with numerous other proteins implicated in USH.14 Usherin likely fulfills important structural and signaling roles in the inner ear and the retina. Rapid and efficient routine testing for USH2A mutations is hampered by the large number of (frequently private) mutations that are scattered throughout this large gene.
In founder populations, such as the French-Canadian population of Quebec and New Brunswick, genetic disorders are often caused by a limited number of mutations in the underlying genes. This has been described for several diseases in the descendants of French settlers in North America.6 French settlement occurred between 1608 and 1759 and mainly in two Canadian regions: along the St Lawrence River (giving rise to the province of Quebec) and in Acadia (today corresponding to Nova Scotia and New Brunswick). Many Acadians were deported to the United States in 1755 for political reasons and they subsequently founded the populations of Cajuns in Louisiana. Some returned later to Maritime Canada. It has been generally accepted that the genetic conditions found in Quebec and Acadia are usually distinct, or they result from different mutations.6 However, we recently showed for the first time that both populations have common ancestors: In both Quebecois and Acadians, deafblindness is due to the same ancestral mutation that is unique to both groups, c.216G>A in the USH1C gene.7 Strikingly, our current results for USH2 strongly support our former findings of genetic linkage of the Acadian and the Quebec population (There are no official numbers about the incidence of USH in Quebec. However, as the families investigated herein were collected over several years and come from different regions of Quebec and the former Acadia, this collective likely is a realistic representation of USH2 patients in these populations).
First, we have identified a predominant USH2A founder mutation (c.4338_4339delCT) in Quebec French-Canadians. In contrast to the situation in other populations, the c.2299delG mutation appears to play a minor role in USH2 in Quebec. Second, the c.4338_4339delCT deletion is present in both Quebecois and Acadians. Of note, only two previous descriptions exist on this mutation, reporting heterozygosity in the presumably same Acadian USH2 family,8, 9 suggesting that this mutation is endemic. As in the case of c.216G>A (USH1C), the c.4338_4339delCT haplotype associated with this USH2A founder mutation is clearly identical in Acadians and Quebecois. This is the second indication that the Acadian and the Quebec population share more founders than has been previously assumed. As a consequence, additional genetic disorders found in both populations are likely based on common founder mutations.
To a large extent, the development of future therapeutic approaches and preventive strategies/disease management in inherited retinal diseases depends on detailed knowledge of the underlying molecular alteration. USH affects the two most important sensory systems (hearing and vision) and results in a major handicap for the patients. Before definitive therapy is at hand, early molecular diagnosis is crucial, as retinal dysfunction succeeds hearing loss with a delay of several years. Once the causative mutation in a hearing-impaired child is known to reside in an USH gene, the parents can consider therapeutic strategies such as cochlear implant at an early point of time. Our current findings provide the molecular tools for simple and efficient diagnostics in USH2 patients from Quebec and former Acadia, with major implications for prognosis and clinical management. Regarding the previous elucidation of USH1 mutations in these populations, USH can now routinely be tested for with minor effort in Canadians and, regarding Louisiana, US Americans with French ancestors.
References
Cohen M, Bitner-Glindzicz M, Luxon L : The changing face of Usher syndrome: clinical implications. Int J Audiol 2007; 46: 82–93.
Adato A, Lefevre G, Delprat B et al: Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum Mol Genet 2005; 14: 3921–3932.
Liu X, Bulgakov OV, Darrow KN et al: Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci USA 2007; 104: 4413–4418.
Ebermann I, Scholl HP, Charbel Issa P et al: A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet 2007; 121: 203–211.
Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ : Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet 2004; 74: 357–366.
Laberge AM, Michaud J, Richter A et al: Population history and its impact on medical genetics in Quebec. Clin Genet 2005; 68: 287–301.
Ebermann I, Lopez I, Bitner-Glindzicz M, Brown C, Koenekoop RK, Bolz HJ : Deafblindness in French Canadians from Quebec: a predominant founder mutation in the USH1C gene provides the first genetic link with the Acadian population. Genome Biol 2007; 8: R47.
Eudy JD, Weston MD, Yao S et al: Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 1998; 280: 1753–1757.
Weston MD, Eudy JD, Fujita S et al: Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet 2000; 66: 1199–1210.
Dreyer B, Tranebjaerg L, Rosenberg T, Weston MD, Kimberling WJ, Nilssen O : Identification of novel USH2A mutations: implications for the structure of USH2A protein. Eur J Hum Genet 2000; 8: 500–506.
Baux D, Larrieu L, Blanchet C et al: Molecular and in silico analyses of the full-length isoform of usherin identify new pathogenic alleles in Usher type II patients. Hum Mutat 2007; 28: 781–789.
Dreyer B, Tranebjaerg L, Brox V et al: A common ancestral origin of the frequent and widespread 2299delG USH2A mutation. Am J Hum Genet 2001; 69: 228–234.
van Wijk E, Pennings RJ, te Brinke H et al: Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet 2004; 74: 738–744.
Kremer H, van Wijk E, Marker T, Wolfrum U, Roepman R : Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet 2006; 15 (Suppl 2): R262–R270.
Acknowledgements
We are indebted to the families who have participated in this study. Supported by grants BO 2954/1-1 (Deutsche Forschungsgemeinschaft), Koeln Fortune Program, grant 113/2004 (Faculty of Medicine, University of Cologne), and Forschung contra Blindheit, Initiative Usher-Syndrom e.V., to HJB, and Foundation Fighting Blindness Canada and Fonds de la Recherche en Sante de Quebec to RKK.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ebermann, I., Koenekoop, R., Lopez, I. et al. An USH2A founder mutation is the major cause of Usher syndrome type 2 in Canadians of French origin and confirms common roots of Quebecois and Acadians. Eur J Hum Genet 17, 80–84 (2009). https://doi.org/10.1038/ejhg.2008.143
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.143
Keywords
This article is cited by
-
Pathogenic variants carrier screening in New Brunswick: Acadians reveal high carrier frequency for multiple genetic disorders
BMC Medical Genomics (2022)
-
Hearing loss in Africa: current genetic profile
Human Genetics (2022)
-
USH2A gene variants cause Keratoconus and Usher syndrome phenotypes in Pakistani families
BMC Ophthalmology (2021)
-
Prevalence and molecular characterization of glucose-6-phosphate dehydrogenase deficiency in the Lue ethnic group of northern Thailand
Scientific Reports (2021)
-
Effect of migration patterns on maternal genetic structure: a case of Tai–Kadai migration from China to Thailand
Journal of Human Genetics (2017)
