
Abstract
Despite the significance of haploinsufficiency in human disease, no systematic study has been reported into the types of genes that are haploinsufficient in human, or into the mechanisms that commonly lead to their deletion and to the expression of the haploinsufficient phenotype. We have applied a rigorous text-searching and database-mining strategy to extract, as comprehensively as possible, from PubMed and OMIM an annotated list of currently known human haploinsufficient genes, including their functions and associated diseases. Gene-set enrichment analysis shows that genes-encoding transcription factors, and genes that function in development, the cell cycle, and nucleic acid metabolism are overrepresented among haploinsufficient genes in human. Many of the phenotypes associated with loss-of-function or deletion of one copy of a haploinsufficient gene describe mental retardation, developmental or metabolic disorders, or tumourigenesis. We also found that haploinsufficient genes are less likely than the complete set of human genes to be situated between pairs of segmental duplications (SDs) that are in close proximity to each other on the same chromosome. Given that SDs can initiate non-allelic homologous recombination (NAHR) and the deletion of adjacent genomic regions, this suggests that the location of haploinsufficient genes between SD pairs, from whence they may suffer intra-genomic rearrangement and loss, is selectively disadvantageous. We describe a custom-made Java visualization tool, HaploGeneMapper, to aid in visualizing the proximity of human haploinsufficient genes to SDs and to enable identification of haploinsufficient genes that are vulnerable to NAHR-mediated deletion.
Similar content being viewed by others

Introduction
Human genetic variation comprises not only single-nucleotide polymorphisms and the variation represented by different alleles of a single genetic locus, but also copy-number variation (CNV) including deletions, insertions, and duplications ranging from 1 kb to 3 Mb in size.1 Segmental duplications (SDs), also known as low-copy repeats (LCRs), can contribute to the generation of some of this CNV. SDs are >5 kb regions in the human genome that share at least 90% sequence identity and that are thought to have originated by duplication.2 Importantly, non-allelic homologous recombination (NAHR) between SDs can result in the deletion or duplication of genetic fragments and/or genes adjacent to them,3 thus altering gene copy number. For some genes, deletion of one functional copy from a diploid genome changes the organism's phenotype to an abnormal or disease state. These genes are called haploinsufficient because a single copy of these genes is insufficient to produce the normal or wild-type phenotype. In the yeast Saccharomyces cerevisiae, where the phenomenon has been relatively well-studied, more than 180 haploinsufficient genes have been identified by fitness profiling of heterozygous deletion strains.4 In human, haploinsufficient genes have recently gained interest because loss of one functional copy has been linked to diseases including neurological disorders5 and mental retardation, for example, 22q11 deletion syndrome,6 Sotos syndrome,7 and Smith–Magenis syndrome.8 Haploinsufficient genes can furthermore influence a person's susceptibility to disease and to the side effects of medications.9
Despite isolated examples describing a physical association of haploinsufficient genes with SDs,10, 11 no systematic study has been conducted to date investigating whether NAHR between SDs could be a common mechanism leading to the deletion of genes and to the expression of haploinsufficient phenotypes. It is also unclear what types of genes are typically haploinsufficient in human. Many experimental approaches for their identification in model systems, such as the fitness profiling mentioned above, cannot be applied in human. We have thus used a rigorous, semi-automated search of the published literature, and genetic databases to summarize, as comprehensively as possible, the current understanding of haploinsufficiency in human. We have retrieved information on haploinsufficient gene loci, their functions, and associated diseases, and have further investigated whether haploinsufficient genes commonly map to regions of SDs. To our knowledge this is the first comprehensive study of human haploinsufficient genes and their genomic proximity to SDs.
Materials and methods
To retrieve published examples of haploinsufficiency, we developed a semi-automated search strategy based on string matching using regular expressions followed by manual curation to search all PubMed and OMIM database entries. In brief, PubMed abstracts were searched for the string ‘haploinsufficien* AND human’ on 12 November 2007; this search returned 1226 records. We then designed a Perl script and regular expressions automatically to extract the gene names corresponding to the haploinsufficient loci described in these 1226 PubMed abstracts. The regular expressions were designed around the following terms, where X is the name of the gene that was extracted: ‘haploinsufficiency for/at/of X’; ‘haploinsufficiency of the X gene’; ‘X haploinsufficiency’; and ‘mutation/s of X leading to haploinsufficiency’. All search results were then compared against known human gene names and symbols to exclude spurious hits. In addition, we searched for haploinsufficient genes in the OMIM database using the search string ‘haploinsufficien* AND human’. All OMIM search results were manually curated to ensure that only true positives and only human gene loci were kept for further analysis. Haploinsufficient genes identified in this manner were stored with their official gene symbol and Entrez gene identifier (ID). Gene functions and associated disorders were manually extracted by searching OMIM, Entrez Gene, and PubMed databases at NCBI (http://www.ncbi.nlm.nih.gov/) using the official gene symbol as search string. Genomic locations of haploinsufficient genes were obtained using the reference assembly of the human genome build 36.2 at NCBI. We also retrieved UniProt/SwissProt accession IDs for all identified haploinsufficient genes using the BioMart tool and official gene symbols in Ensembl (http://www.ensembl.org/index.html/). Gene-set enrichment analysis was performed using the GOstat browser (http://gostat.wehi.edu.au/)12 and the complete set of annotated gene products in the GO gene-associations database of the European Bioinformatics Institute (GOA Human 56.0). This gene-associations file contained 33 731 gene products of which 27 018 had GO terms associated with them. We thus compared the GO annotations of all gene products encoded by haploinsufficient genes to GO annotations of the complete set of human gene products. In addition, we performed a second gene-set enrichment analysis using only gene products of OMIM disease-related genes and haploinsufficient genes as a reference set instead of the complete set of human gene annotations in GOA Human. We thus obtained all OMIM entries with the ‘+’ flag, which refer to entries with a known gene sequence and phenotype and which are usually associated with disease (http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM). This resulted in 388 OMIM disease-related gene entries corresponding to 391 Entrez Gene IDs and 381 UniProt IDs. These 388 OMIM entries were combined with the 299 haploinsufficient gene loci identified in this study to produce the reference set for the second gene-set enrichment analysis. This new reference set contained a total of 638 genes. We then compared the annotated functions of gene products encoded by haploinsufficient genes to the new reference set containing a total of 638 haploinsufficient and OMIM disease-related genes. In both gene-set enrichment analyses, Benjamini–Hochberg's method for multiple testing was used to adjust the false discovery rate,13 and gene functions with P<10e-5 were considered to be significantly over- or underrepresented.
To investigate the genomic proximity of the identified haploinsufficient genes to SDs, we downloaded the latest human SD data from the Human Genome Segmental Duplication Database (http://projects.tcag.ca/humandup/).2 These analyses are based on the Human Genome May 2004 Assembly, Human Build 35. As the frequency of NAHR between SDs increases as the distance between SDs decreases,14, 15 we identified for each human haploinsufficient gene the nearest SD pair flanking it, and determined the distance between the two paired SDs (Figure 1). We then compared the inter-SD distance for pairs enclosing haploinsufficient genes, to the inter-SD distance for pairs enclosing all remaining human genes. Total human gene count (31 210) and the genomic location of these gene loci were based on the Ensembl v47 release. A Java application, HaploGeneMapper, was developed to visualize the proximity of human haploinsufficient genes to SDs. Each SD displayed in HaploGeneMapper is identified by the duplicon ID taken directly from the Human Genome Segmental Duplication Database. These IDs are of the type DP_A_B_C, where A is the chromosome of the first duplicon in a SD pair, B is the chromosome of the second duplicon in a SD pair, and C is a number assigned consecutively by the position of the first duplicon on the A chromosome. Human gene IDs displayed in HaploGeneMapper are taken from Ensembl v47.

Calculation of distances between pairs of segmental duplications (SDs). Grey shaded boxes represent SDs. Pairs of SDs, which are thought to have originated by duplication, are labelled with the same letters, here A and A* and B and B*. Such SD pairs can be subject to non-allelic homologous recombination (NAHR) and cause deletion of adjacent regions. Another SD mapping to this region, C, does not have a corresponding SD on this chromosome segment. H1 and H2 represent haploinsufficient genes identified in this study. There are also two other human genes on this chromosome segment, N1 and N2, which have not been described as haploinsufficient. For each haploinsufficient gene, we identified the nearest SD pair enclosing it and determined the genomic distance dx between the SDs in the pair. Such distances reflect the probability for NAHR, with distances less than 10 Mb showing the greatest incidence of NAHR. In this example, haploinsufficient gene H1 is enclosed by SD pairs A and B, but since dB is smaller than dA, SD pair B is taken as the closest SD pair flanking H1. In contrast, haploinsufficient gene H2 is flanked by SD pair A, and dA is taken as the distance between SDs for this gene. We then compared the distance between corresponding SDs for SD pairs enclosing haploinsufficient genes to those enclosing other human genes.
Results
Using a rigorous search of the published literature and the OMIM genetic database, we identified 299 human gene loci previously described as haploinsufficient. Of those, 88 were retrieved only by searching the OMIM database, 94 only by searching the published literature in PubMed, and 117 by using both search strategies. The complete list of human haploinsufficient genes, including PubMed references and OMIM entries used for their identification, is available as Supplementary Table 1. Of these 299 identified human haploinsufficient genes, gene function information was associated with 281. Gene-set enrichment analysis showed that several functional categories were significantly enriched among human haploinsufficient genes, including various functions in development, the cell cycle, nucleic acid metabolism, and transcription (Table 1). Only GO terms that were enriched regardless of whether the complete set of human GOA annotations, or the subset of OMIM disease-related gene annotations, was used as a reference set, are shown in Table 1. The complete list of enriched gene ontology terms using all human GOA annotations as a reference set is available as Supplementary Table 2. Most of the identified haploinsufficient genes have already been associated with human disease or disease susceptibility. For example, 21% of human haploinsufficient genes have been associated with cancer and tumourigenesis, 15% with mental retardation, 6% with neurological disorders, 4% with growth retardation, and 4% with developmental disorders (Supplementary Table 1). The remaining 50% have been associated with a variety of other disorders including immunodeficiency, metabolic disorders, kidney disease, limb malformation, and infertility (Supplementary Table 1).
Of the 299 human haploinsufficient genes, 177 (59%) were located between pairs of SDs on the same chromosome, and a further 11 (4%) were located inside a SD. In comparison, 18 925 (61%) of the remaining human genes were located between SD pairs and 2766 (9%) were located inside a SD. Thus, on a chromosome-wide scale, haploinsufficient genes are located between SDs with a similar frequency as the remaining human genes, but a smaller percentage of haploinsufficient genes is located directly inside a SD. The distance between corresponding SDs on the same chromosome ranged from 2.7 kb to 246 Mb. As the distance between SD pairs decreased, haploinsufficient genes were consistently less likely than the remainder of human genes to be situated between corresponding SDs, and this is especially the case for SD pairs that are in very close proximity (less than 10 Mb) to each other (Figure 2). For example, only 29% of haploinsufficient genes were located between SD pairs less than 10 Mb apart compared to 36% of the remaining human genes (Figure 2), and only 11% were located between SD pairs less than 1 Mb apart compared to 20% of the remaining human genes (Figure 2).

The percentage of haploinsufficient genes situated between pairs of segmental duplications (SDs). The distance between pairs of SDs, which may be subject to non-allelic homologous recombination, is given in Mb on the x axis. These distances were grouped into bins of 10 Mb, with the bin labels describing the maximum distance found in the bin category. The primary y axis and the solid line data refer to the percentage of genes that are situated between SD pairs for a given SD pair distance. Grey triangles represent human haploinsufficient genes identified in this study, while black squares represent the remaining human genes. The secondary y axis and the dotted line data refer to the cumulative percentage of genes that are situated between SD pairs for a given SD pair distance. SD pair distances ranged from less than 10–250 Mb. Haploinsufficient genes are less likely than the remaining human genes to be situated between physically proximate SD pairs, especially those less than 10 Mb distance apart. The grey shaded area is expanded in the insert and represents those genes situated between SD pairs that are less than 10 Mb distance apart.
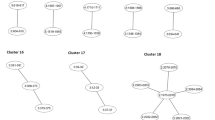
We developed a Java application, HaploGeneMapper, to visualize the genomic proximity of haploinsufficient genes to SDs on a chromosome-by-chromosome basis (Figure 3a shows the screenshot for human chromosome 17). Pairs of SDs, which may be subject to NAHR, can be highlighted using the cursor. Similarly, gene and SD names can be displayed by hovering the cursor over them. If required, the user can zoom into a section of the selected chromosome by providing the start and end coordinates of the desired region. An executable jar distribution is available at http://haplogenemapper.sourceforge.net/. HaploGeneMapper is platform-independent, but requires Java Runtime Environment 1.6 or later. The input files consist of the list of human haploinsufficient genes compiled in this study and the genomic coordinates of human SDs (taken from Cheung et al2), both of which are available at the above-mentioned website. HaploGeneMapper allows any researcher to browse the human genome with ease to identify whether a particular human haploinsufficient gene of interest is in close proximity to SDs. The application further allows the user quickly to gauge the distances between corresponding SDs that bear relevance for the probability of NAHR between them. Using this visualization tool we observed that human chromosome 17 is particularly enriched for haploinsufficient genes that are located between physically proximate SD pairs (Figure 3a). These include, for example, the haploinsufficient genes RAI1, COPS3, TOP3A, and SHMT1 located between eight corresponding SD pairs (Figure 3b). These SDs are separated by only 1.5 Mb and share >93% sequence identity, making them particularly susceptible to NAHR. So far as we can determine, other genes situated between these SD pairs, such as NT5M and ATPAF2, have not been described as haploinsufficient. The list of haploinsufficient genes presented in Supplementary Table 1 includes only those genes that have been already described as haploinsufficient, and not other genes, such as ATPAF2, which are simply located in the same genomic context or microdeletion region as known haploinsufficient genes. However, we expect that, with the availability of more data, some of these other genes in common microdeletion regions may also be identified as haploinsufficient in the future.

HaploGeneMapper, a custom JAVA application to illustrate the genomic proximity of haploinsufficient genes to segmental duplications (SDs). A screenshot of the data for human chromosome 17 is shown in (a). Corresponding pairs of SDs can be highlighted in the same colour by hovering the cursor over the SD of interest. The position of human haploinsufficient genes relative to SD pairs can be easily examined on a chromosome-by-chromosome basis. Chromosome 17 is particularly enriched for haploinsufficient genes that are situated between SD pairs that are in close proximity to each other on the same chromosome. For example, a region corresponding to 17p11.2, indicated by the blue dotted box and expanded below the screenshot, contains four haploinsufficient genes. These are RAI1, COPS3, TOP3A, and SHMT1 and are situated between four pairs of physically proximate SDs, labelled A and A* to D and D* (these correspond to the SDs DC_17_17_6383, DC_17_17_6499, DC_17_17_6369, DC_17_17_6541, DC_17_17_6366, DC_17_17_6551, DC_17_17_6360, and DC_17_17_6560 in the Human Segmental Duplications Database) (b). Each SD is only 1.6–2 Mb distant from its corresponding partner with which it shares at least 93% sequence identity. These physically proximate SD pairs of high sequence identity thus have the potential to initiate non-allelic homologous recombination. Deletion of this genomic region and the haploinsufficient genes RAI1, COPS3, TOP3A, and SHMT1 is associated with Smith–Magenis syndrome, a contiguous gene syndrome that includes speech delay, mental retardation, and behavioural problems. Smith–Magenis Syndrome Repeat Gene Clusters including a distal (SMS-REPD), middle (SMS-REPM), and proximal (SMS–REPP) are three repeat gene clusters previously linked to NAHR and genomic rearrangements in this region.10 Their approximate location with respect to the SD pairs described above is indicated by the grey shaded boxes. Other genes that also map to this genomic region but do not appear to be haploinsufficient include NT5M and ATPAF2.
Discussion
Using a combination of text-searching and database-mining strategies, we have identified in PubMed and OMIM 299 haploinsufficient genes in human, many of which function in development, the cell cycle, nucleic acid metabolism or transcription. Deletion of one copy of these haploinsufficient genes is associated most frequently with tumourigenesis, developmental disorders, and mental retardation. We found that haploinsufficient genes are less likely than the remaining human genes to be located between pairs of SDs that are in close proximity, especially less than 10 Mb, to each other on the same chromosome. We have made publicly available a Java application tool, HaploGeneMapper, which allows the physical proximity of haploinsufficient genes to SDs to be visualized easily.
Theoretically, all haploinsufficient genes represented in the OMIM database should have a PubMed reference, and thus should also have been retrieved by our PubMed text-searching strategy. However, based on the results from the OMIM search, we missed approximately 26% of haploinsufficient genes in the PubMed search. We also tried other search parameters, but these returned a very large number of results with many spurious hits and false positives. We were thus unable to increase the sensitivity of our text-searching strategy without also increasing the number of false positive hits to a number that precluded manual assessment of all results. In order to ensure high quality of the results reported here, we retained the existing strategy, acknowledging that the true number of haploinsufficient genes represented in PubMed is probably greater than discussed here.
What types of genes are typically dosage sensitive, including haploinsufficient, and why, has been a subject of lengthy debate. Extrapolating from selected examples, previous studies have suggested that proteins with structural, regulatory, mechanochemical, and other non-enzymatic functions are most likely to underlie the haploinsufficient phenotype.16, 17, 18, 19 To assess what functional categories are enriched among haploinsufficient genes, we compared the annotated functions of gene products encoded by haploinsufficient genes to those of gene products encoded by disease-related genes found in OMIM, as well as to the complete set of human GOA annotations. Despite the use of two different reference sets during gene-set enrichment analysis, we obtained similar, significantly enriched GO terms. It is likely that the number of disease-related genes in OMIM currently underestimates the true number of human disease-related genes. On the other hand, using the full set of human gene products for comparison with human haploinsufficient genes takes no account of the types of genes that are disease-related and may thus be haploinsufficient. For these reasons, we present here only those GO terms that were found to be significantly enriched in both the analyses. One of the most significantly enriched GO terms in our analyses was the term ‘transcription factor activity’, which showed a fivefold enrichment among haploinsufficient genes. Our results, based on a far broader representation (299 genes) than previous studies, thus support the notion that haploinsufficient genes preferentially encode regulatory proteins and transcription factors. As a result, many haploinsufficient genes are expected to affect gene expression20 and link to disease and disease susceptibility by altering genetic regulatory networks.
In some cases, it is possible to relate the haploinsufficient phenotype directly to the function of the haploinsufficient locus. For example, deletion of gene copies for FOXP2, which is involved in the development of language skills, has been shown to lead to speech and language impairment.21 Similarly, deletion of tumour suppressor genes, for example, FBW7, has been associated with tumourigenesis.22 Other disorders are caused by alterations in copy number at several haploinsufficient gene loci. For example, mental retardation associated with the 22q11 deletion syndrome is due to the combined effects of the haploinsufficient genes PRODH23 and TBX1.24 Our study has found that many haploinsufficient phenotypes describe developmental disorders and mental retardation, consistent with our finding that haploinsufficient genes are enriched in regulatory and developmental gene functions.
SDs appear to be important in primate evolution and in shaping human genetic variation.3 The high sequence identity between SDs can initiate NAHR, which has been recognized as a major cause of gene deletions, duplications, and inversions associated with genomic disorders.3 Such rearrangements are particularly frequent between SDs that share greater than 95% sequence identity, are more than 10 kb in length, and are separated by only 20 kb to 10 Mb.14 We have presented here an example in which four haploinsufficient genes, RAI1, COPS3, TOP3A, and SHMT1, are located between a series of SD pairs of high sequence similarity that are also in close proximity to each other on the same chromosome. The regions encompassing these SD pairs have been termed ‘Smith–Magenis Syndrome Repeat Gene Clusters’ (SMS-REPs) and include a distal (SMS-REPD), middle (SMS-REPM), and proximal (SMS-REPP) gene cluster.10 Deletions of RAI1, TOP3A, and SHMT1 associated with Smith–Magenis syndrome have already been linked to NAHR between SMS-REPD, SMS-REPM, and SMS-REPP.10, 25 Hence, other genes within this region would also be vulnerable to deletion by NAHR, but many of these other genes, such as NT5M and ATPAF2 (which encode a nucleotidase and an ATP-synthase assembly factor, respectively) have not been identified as haploinsufficient. Genes that encode enzymes have been previously described as less likely to be haploinsufficient.18 Why deletion of ATPAF2, which also maps to this deletion region, does not cause a significant change in phenotype is currently unclear.
We have found that haploinsufficient genes are less likely than the remaining human genes to be situated between physically proximate SD pairs. This bias may appear because genomic proximity of haploinsufficient genes to SDs is selectively disadvantageous. However, we cannot exclude the possibility that some of this bias may be due to genomic regions rich in SDs being genetically poorly characterized. It is possible, for example, that the complexity of SD-rich regions has precluded them from being well characterized and that, as a result, many genes located in these regions have not yet been associated with genetic disorders and haploinsufficiency. If this is the case, the true number of haploinsufficient genes located in such regions would be underestimated.
HaploGeneMapper has proven to be a useful tool to identify genomic regions that are rich in SDs and to examine the physical distance between corresponding SD pairs as well as to locate the genes that are located in these regions. Although it is possible to design custom tracks in the UCSC browser or to browse the location of SDs in GBrowse of the Human Genome Segmental Duplication Database, these former applications lack a number of features we considered essential for this study. For example, HaploGeneMapper easily displays the SD data for complete human chromosomes and thus provides a large-scale overview we have found useful in identifying genomic regions particularly rich in SDs. Using this approach, we immediately found the genomic region containing the Smith–Magenis Syndrome repeat clusters on chromosome 17 to be enriched for SDs, resulting in our choice of this genomic region as a case study. In addition, corresponding SDs that may be subject to NAHR are highlighted when hovering the cursor over an SD. This feature of HaploGeneMapper facilitates the identification of SD pairs that are in close proximity to each other as well as the genes that are flanked by these SDs. This information can be useful in judging whether a particular gene of interest could be subject to NAHR-mediated deletion.
Haploinsufficiency is not specific to human, but has also been described in yeast.4 We were thus interested in whether human haploinsufficient genes identified in this study corresponded to haploinsufficient genes identified experimentally in yeast.4 To map orthologs between human and yeast we used the Ensembl Compara database (http://www.ensembl.org/index.html/). Only two of the 299 human haploinsufficient genes were orthologous to genes identified experimentally as haploinsufficient in yeast: the gene RPS19-encoding ribosomal protein S19 corresponding to yeast YOL121C, and the gene RPS4X-encoding ribosomal protein S4 (X linked) corresponding to yeast YJR145C and YHR203C. The limited overlap between human and yeast haploinsufficient genes suggests that either the sampling was too limited in extent, or yeast and human have fundamentally different gene regulatory and gene interaction networks. Both RPS19 and RPS4X genes encode proteins that, in function and sequence, are highly conserved across eukaryotes.
Many experimental approaches for identifying genes as haploinsufficient in model systems are not applicable to studies in human. Our current understanding of human haploinsufficient genes thus remains limited, and we do not know which genes are definitely not haploinsufficient. It is also possible that genes are haploinsufficient in one genomic or environmental context, but not in another. For these reasons, it is currently unclear how many haploinsufficient genes are yet to be identified in the human genome. We have attempted to compile, as comprehensively as possible, a list of human haploinsufficient genes using a combination of text-searching and database-mining strategies. With the availability of additional human genome sequences in the near future, it will be possible to identify further regions subject to CNV, new haploinsufficient genes that map to these regions, and their contribution to human disease and disease susceptibility. It has been suggested that CNV and genomic rearrangements have a greater effect on human phenotypes than do single-nucleotide polymorphisms. Understanding this variation, and the mechanisms which create it, will thus be critical to advance our understanding of human disease and disease susceptibility.
References
Feuk L, Carson AR, Scherer SW : Structural variation in the human genome. Nat Rev Genet 2006; 7: 85–97.
Cheung J, Estivill X, Khaja R et al: Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence. Genome Biol 2003; 4: 1–10.
Bailey JA, Eichler EE : Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet 2006; 7: 552–564.
Deutschbauer AM, Jaramillo DF, Proctor M et al: Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics 2005; 169: 1915–1925.
Sisodiya SM, Ragge NK, Cavalleri GL et al: Role of SOX2 mutations in human hippocampal malformations and epilepsy. Epilepsia 2006; 47: 534–542.
Meechan DW, Maynard TM, Gopalakrishna D, Wu Y, Lamantia AS : When half is not enough: gene expression and dosage in the 22q11 deletion syndrome. Gene Expr 2007; 13: 299–310.
Kurotaki N, Stankiewicz P, Wakui K, Niikawa N, Lupski JR : Sotos syndrome common deletion is mediated by directly oriented subunits within inverted Sos-REP low-copy repeats. Hum Mol Genet 2005; 14: 535–542.
Elsea SH, Girirajan S : Smith–Magenis syndrome. Eur J Hum Genet 2008; 16: 412–421.
Cohen J : DNA duplications and deletions help determine health. Science 2007; 317: 1315–1317.
Chen K-S, Manian P, Koeuth T et al: Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet 1997; 17: 154–163.
Venturin M, Gervasini C, Orzan F et al: Evidence for non-homologous end joining and non-allelic homologous recombination in atypical NF1 microdeletions. Hum Genet 2004; 115: 69–80.
Beissbarth T, Speed TP : GOstat: find statistically overrepresented gene ontologies within a group of genes. Bioinformatics 2004; 20: 1464–1465.
Benjamini Y, Hochberg Y : Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol) 1995; 57: 289–300.
Lacroix MH, Oparina NY, Mashkova TD : Segmental duplications in the human genome. Mol Biol 2003; 37: 186–193.
Stankiewicz P, Lupski JR : Genome architecture, rearrangements and genomic disorders. Trends Genet 2002; 18: 74–82.
Fisher E, Scambler P : Human haploinsufficiency – one for sorrow, two for joy. Nat Genet 1994; 7: 5–7.
Veitia AR : Exploring the etiology of haploinsufficiency. BioEssays 2002; 24: 175–184.
Kondrashov FA, Koonin EV : A common framework for understanding the origin of genetic dominance and evolutionary fates of gene duplications. Trends Genet 2004; 20: 287–290.
Kacser H, Burns JA : The molecular basis of dominance. Genetics 1981; 97: 639–666.
McCarroll SA, Hadnott TN, Perry GH et al: Common deletion polymorphisms in the human genome. Nat Genet 2006; 38: 86–92.
Zeesman SNM, Teshima I, Roberts W et al: Speech and language impairment and oromotor dyspraxia due to deletion of 7q31 that involves FOXP2. Am J Med Genet A 2006; 140A: 509–514.
Welcker M, Clurman BE : FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008; 8: 83–93.
Weksberg R, Stachon A, Squire J et al: Molecular characterization of deletion breakpoints in adults with 22q11 deletion syndrome. Hum Genet 2007; 120: 837–845.
Arinami T : Analyses of the associations between the genes of 22q11 deletion syndrome and schizophrenia. J Hum Genet 2006; 51: 1037–1045.
Vlangos CN, Yim DKC, Elsea SH : Refinement of the Smith–Magenis syndrome critical region to ∼950 kb and assessment of 17p11.2 deletions. Are all deletions created equally? Mol Genet Metabol 2003; 79: 134–141.
Acknowledgements
We thank Dr Steve Scherer (Sick Kids’ Hospital, Toronto) for permission to use data on predicted segmental duplications in human. We acknowledge the support of Australian Research Council grant CE0348221 (ARC Centre of Excellence in Bioinformatics) and of an Australian Development Scholarship (to VTD).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Rights and permissions
About this article
Cite this article
Dang, V., Kassahn, K., Marcos, A. et al. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur J Hum Genet 16, 1350–1357 (2008). https://doi.org/10.1038/ejhg.2008.111
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.111
Keywords
This article is cited by
-
Phase separation as a possible mechanism for dosage sensitivity
Genome Biology (2024)
-
An unsupervised deep learning framework for predicting human essential genes from population and functional genomic data
BMC Bioinformatics (2023)
-
A fine balancing act: how epitranscriptome regulates dosage compensation in mammals
Nature Structural & Molecular Biology (2023)
-
Clinical implementation of RNA sequencing for Mendelian disease diagnostics
Genome Medicine (2022)
-
Augmenting and directing long-range CRISPR-mediated activation in human cells
Nature Methods (2021)
