
Abstract
ASXL1 mutations are frequently found in hematological tumors, and loss of Asxl1 promotes myeloid transformation in mice. Here we present data supporting a role for an ASXL1-BAP1 complex in the deubiquitylation of mono-ubiquitylated lysine 119 on Histone H2A (H2AK119ub1) in vivo. The Polycomb group proteins control the expression of the INK4B-ARF-INK4A locus during normal development, in part through catalyzing mono-ubiquitylation of H2AK119. Since the activation of the locus INK4B-ARF-INK4A plays a fail-safe mechanism protecting against tumorigenesis, we investigated whether ASXL1-dependent H2A deubiquitylation plays a role in its activation. Interestingly, we found that ASXL1 is specifically required for the increased expression of p15INK4B in response to both oncogenic signaling and extrinsic anti-proliferative signals. Since we found that ASXL1 and BAP1 both are enriched at the INK4B locus, our results suggest that activation of the INK4B locus requires ASXL1/BAP1-mediated deubiquitylation of H2AK119ub1. Consistently, our results show that ASXL1 mutations are associated with lower expression levels of p15INK4B and a proliferative advantage of hematopoietic progenitors in primary bone marrow cells, and that depletion of ASXL1 in multiple cell lines results in resistance to growth inhibitory signals. Taken together, this study links ASXL1-mediated H2A deubiquitylation and transcriptional activation of INK4B expression to its tumor suppressor functions.
Similar content being viewed by others

Introduction
The INK4B-ARF-INK4A locus plays important roles in the cellular defense against tumorigenesis, and it is frequently deleted, mutated or methylated in various primary tumors1,2. The locus is tightly controlled and is kept silent during embryogenesis and in normal proliferating cells. The Polycomb group proteins (PcG)3,4 are essential for maintaining the locus in a repressed state. These proteins form part of several different complexes of which the two most studied are polycomb repressive complex 2 (PRC2) and PRC1. These complexes impose their repressive functions, in part, through catalyzing histone modifications: PRC2 catalyzes tri-methylation of histone H3 Lys 27 (H3K27me3) and PRC1 catalyzes mono-ubiquitinylation of H2AK119 (H2AK119ub1)5,6. The activation of the INK4B-ARF-INK4A locus by oncogenes or stress-induced signals leads to cellular senescence, thereby limiting the proliferation of the damaged cells that are at risk of neoplastic transformation7,8,9. However, the products of the locus — p15INK4B, p14ARF, and p16INK4A— are not redundant and play independent roles in restricting proliferation1. The INK4B locus is particularly prone to induction by anti-proliferative signals during differentiation and development10,11,12. Moreover, co-deletion of Ink4b with Ink4a-Arf in mice results in a broader spectrum of tumors compared with individual genetic deletion13. The full understanding of the mechanisms leading to their separate or coordinate activation of the locus is still lacking.
Asx is a relatively poorly characterized gene belonging to the enhancer of Trithorax and Polycomb (ETP) group and its deletion causes both posterior and anterior transformation in Drosophila14. Scheuermann et al.15 showed that Asx is in a complex with Calypso (the Drosophila homolog of human BAP1), an ubiquitin carboxy-terminal hydrolase that deubiquitylates H2AK119ub1. Drosophila with Asx or Calypso mutations showed a strong increase in the levels of H2AK119ub1, but surprisingly this increase was correlated with derepression of PcG-targeted Hox genes. Therefore, this complex was named as polycomb repressive deubiquitinase complex (PR-DUB)15. However, the mechanism by which Asx mutations lead to the derepression of Hox genes is still uncertain.
ASXL1, one of the mammalian Asx homologs, is required for proper axial patterning in mice and both silencing and activation of Hox genes16. ASXL1 mutations are frequently found in diverse human tumors such as hematological malignancies17,18,19,20, breast cancers21 and prostate cancers22. ASXL1 mutations in patients with myelodysplastic syndrome (MDS) and chronic myelomonocytic leukemia (CMML) usually correlate with acute transformation and worse prognosis23,24,25. Recently, mouse genetic studies confirmed that loss of function of Asxl1 leads to MDS-like defects26,27,28, and that loss of Asxl1 in combination with activated N-Ras or loss of Tet2 increases the severity of the hematological malignancy27,28. Mechanistically, Abdel-Wahab et al.29 showed that ASXL1, as a tumor suppressor, prevents the expression of oncogenes like HOXA9 by association with PRC2. However, a role for the catalytic function of the ASXL1 and BAP1 containing complex in activating transcription has not been described.
In this study, we have addressed whether the catalytic function of the ASXL1-BAP1 complex plays an active role in antagonizing PcG functions in mammals, and whether this function could explain a role for ASXL1-BAP1 in tumor suppression. We confirmed that mammalian ASXL1 interacts with BAP1 and is essential for H2A deubiquitylation in vitro and in vivo. Different from the categorization as a PR-DUB15, we provide direct evidence that the complex is essential for the activation of INK4B expression by a mechanism involving the removal of the transcriptionally repressive mark H2AK119ub1 from the INK4B locus in both human and mouse cells. Our studies demonstrate an important mechanism for ASXL1 acting as a tumor suppressor whose loss obviates intrinsic or extrinsic anti-proliferative programs.
Results
ASXL1 forms an H2A deubiquitylation complex by interacting with BAP1
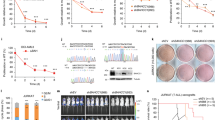
ASXL1 has been found to interact with BAP115 and PRC229. To systematically identify proteins binding to mammalian ASXL1, we generated human 293 cells with inducible expression of FLAG-HA-tagged ASXL1 (FH-ASXL1). ASXL1 and interacting proteins were purified from nuclear extracts by FLAG- followed by HA-affinity chromatography. Subsequently, the proteins associated with ASXL1 were identified by mass spectrometry (MS) analysis. HCF1, OGT, FOXK1, FOXK2 and BAP1 were highly enriched in the affinity purification of ASXL1-associated proteins (Figure 1A). Notably, we did not detect any peptides for PRC2 members. Similar results were obtained from purification of the Asxl1 complex in murine embryonic stem cells (data not shown). The interactions were validated by independent immunoprecipitation (IP) assays followed by western blotting (Supplementary information, Figure S1A). Moreover, we performed reciprocal endogenous co-IP assays using total lysates prepared from mouse myeloblastic leukemia cell line M1, with antibodies specific for ASXL1, BAP1 and SUZ12. As shown in Figure 1B, ASXL1 and BAP1 interact with each other. However, no association was detected between the PRC2 components EZH2 and SUZ12 and ASXL1 or BAP1. To test whether PRC2 components co-purify with BAP1 and HCF1, we generated 293 cells with inducible expression of FLAG-HA-tagged BAP1 (FH-BAP1) and FH-HCF1. We used these cell lines for tandem affinity purification to identify BAP1- and HCF1-associated proteins. As shown in Supplementary information, Figure S1B, S1C and Figure 1C, ASXL, BAP1, HCF1, OGT and FOXK1/2 co-purify with each other, however, we were not able to detect any trace of PRC2 members associated with BAP1 and HCF1, similar to what we found for ASXL1. Consistent with these results, ASXL1 and BAP1 co-elute with HCF1 in high molecular weight (MW) fractions in 293 cells (around 2 000 KDa), which is different from the distribution of the PcG proteins (RING1B and EZH2) that mainly elute in the lower MW fractions (Supplementary information, Figure S1D). Therefore, human ASXL1 forms an endogenous complex similar to PR-DUB.

ASXL1 forms an H2A deubiquitylation complex by interacting with BAP1. (A) Summary of the peptides identified by mass spectrometry from a FLAG-HA tandem purification of FLAG-HA-ASXL1 expressed in Flp-In T-REX 293 cells. Mass (Da), observed number of peptides and the score obtained by Mascot database searching (Matrix Science, UK) were indicated for each protein. (B) Co-IP of endogenous ASXL1 and BAP1 proteins in myeloblastic leukemia cells M1. The input lanes contain 5% of the amount used for IP. The ASXL1 and BAP1 mouse monoclonal antibody, SUZ12 rabbit monoclonal antibody were used for IP. HA was used as negative controls in IP. (C) Summary of the peptides that are in common identified by MS from the purifications of FH-ASXL1, FH-BAP1 and FH-HCF1 expressing 293 cells. (D) ASXL1 stimulates H2A deubiquitylation activity of BAP1 in vitro. In vitro deubiquitylation assays were performed using the indicated amounts of FLAG-tagged BAP1 recombinant protein with or without FLAG-tagged ASXL1. Nucleosomes purified from HeLa cells were used as substrates. After incubation for 1 h, western blot analyses were carried out with the indicated antibodies. (E) ASXL1 stimulates H2A deubiquitylation activity in vivo. Levels of H2AK119ub1 and H3K27me3 in ASXL1-mutated NOMO1 cells with or without ectopic expression of ASXL1. Histone H3 was used as a loading control.
PR-DUB in Drosophila was found to specifically remove ubiquitin from H2A in vitro and in vivo15, and the human homologs were shown to possess the same activity in vitro. To confirm the biochemical activity, we tested the in vitro deubiquitylation activity of full-length recombinant proteins of human ASXL1 and BAP1 using HeLa-derived oligonucleosomes as substrates. As shown in Figure 1D, even high amounts of the deubiquitinase BAP1 did not show any activity on H2AK119ub1 in the absence of ASXL1. However, addition of ASXL1 promoted dose-dependent deubiquitylation of H2A by BAP1, demonstrating the requirement of ASXL1 for BAP1-catalyzed H2A deubiquitylation in vitro. Noteworthy, we did not detect any activity on H2BK120 ubiquitinylation even using high doses of both proteins (upper panel, Figure 1D). Moreover, the catalytic activity relied on BAP1, as a point mutation in the catalytic domain of BAP1 (BAP1-A95D) completely abolished the effect on H2AK119ub1 (lower panel, Figure 1D).
Consistent with the in vitro activity, the H2AK119ub1 levels were decreased when ASXL1 was ectopically expressed in NOMO1. The expression of ASXL1 did not lead to detectable changes in H3K27me3 levels (Figure 1E). Therefore, in agreement with previous publications, our data show that a complex containing ASXL1 and BAP1 possesses H2A deubiquitylation activity in vitro and in vivo.
ASXL1 expression is induced by oncogenic BRAF and required for the activation of INK4B expression
Given that the PcG proteins are required for maintaining the repression of INK4B-ARF-INK4A locus, we tested whether ASXL1 antagonizes PcG repressive functions in this context. We took advantage of a well-characterized system to study oncogene-induced senescence in which human diploid fibroblasts, TIG3 contain an inducible form of the BRAF oncogene (ΔBRAF-ER30). Activation of BRAF by the treatment with 4-hydroxytamoxifen (4-OHT) leads to the activation of the INK4B-ARF-INK4A locus and cellular senescence (Figure 2A). Using this system, we examined whether ASXL1 would be differentially expressed in response to oncogene activation. As measured by real-time quantitative PCR (qPCR; Figure 2B) and western blot (Figure 2C), the activation of BRAF led to a significant and rapid increase in ASXL1 expression levels, but not in BAP1 levels (Figure 2B and 2C). The increase in ASXL1 expression appeared to slightly precede the increase in the expression of p15INK4B, p16INK4A and p14ARF (Figure 2B and 2C).

Depletion of ASXL1 attenuates oncogenic BRAF-induced p15INK4B expression. (A) Activation of BRAF in TIG3 ΔBRAF-ER cells by 4-OHT induces senescence. (B, C) Expression levels of ASXL1, p15INK4B, p16INK4A and p14ARF in TIG3 ΔBRAF-ER at the indicated times following 4-OHT treatment determined by RT-qPCR (B) and western blot analysis (C). (D) Real-time qPCR analyses to determine the relative mRNA levels of ASXL1, INK4B, INK4A and ARF in TIG3 ΔBRAF-ER cells transduced with two different shRNA constructs targeting ASXL1 (shASXL1-B and shASXL1-C) or scramble shRNA (Scr) at different times of 4-OHT treatment. The error bars denote SD, n = 3.
Subsequently, we tested whether the activation of the INK4B-ARF-INK4A locus was dependent on ASXL1 by downregulating ASXL1 expression using shRNAs towards different regions of ASXL1 (shASXL1-B and -C, Supplementary information, Figure S2A and S2B, Figure 2D). Interestingly, the expression of INK4B, but not INK4A or ARF, failed to be efficiently induced upon depletion of ASXL1 (Figure 2D). Taken together, these results show that ASXL1 is required for the efficient transcriptional activation of INK4B in response to oncogene activation.
ASXL1 is required for the increased INK4B expression in response to anti-proliferative signals
In addition to cell-autonomous oncogenic pathways, INK4B expression is increased in response to extrinsic anti-proliferative signals. For instance, INK4B expression is upregulated in epithelial cells treated with transforming growth factor β (TGFβ), and p15INK4B is required for TGFβ-mediated cell cycle arrest11. INK4B expression is also increased in myeloid progenitor cells by interleukin-6 (IL6), interferon β (IFNβ) and lipopolysaccharide (LPS) and is involved in the physiologic control of hematopoietic cell growth31.
To investigate whether ASXL1 could also have a role in regulating INK4B expression in response to these physiological anti-proliferative signals, we used immortalized human epithelial cells (HaCaT)11 and murine leukemia cells M131 as model systems. As shown by immunostaining, short-term treatment of HaCaT cells with TGFβ induced a gradual increase in the expression of p15INK4B (Figure 3A). Moreover, the increase of p15INK4B mRNA and protein levels was highly dependent on the expression of ASXL1 (Figure 3B and 3C). In contrast, the expression of the other two tumor suppressor genes INK4A and KIP1 was not affected by either TGFβ treatment or depletion of ASXL1 (Figure 3B and 3C). To exclude the possibility of off-target effects, we re-introduced shRNA-resistant mAsxl1 into the ASXL1-depleted HaCaT cells (Supplementary information, Figure S2C). As measured by real-time qPCR, the INK4B induction efficiency was restored by the rescued mAsxl1 expression (Supplementary information, Figure S2D).

Loss of ASXL1 attenuates extrinsic signal-induced p15INK4B expression. (A) Immunofluorescence staining of p15INK4B in HaCaT cells at the indicated times following TGFβ treatment. Scale bar, 30 μm. (B) Real-time qPCR analyses to determine the relative mRNA levels of ASXL1, INK4B, INK4A and ARF in control (Scr) or ASXL1-depleted (shASXL1-B and shASXL1-C) HaCaT cells at different times of TGFβ treatment. The error bars denote SD, n = 3. (C) Western blot analysis of ASXL1, ASXL1p16INK4A and p15INK4B in control (Scr) or ASXL1-depleted (shASXL1-B) HaCaT cells with or without 24 h of TGFβ treatment. β-actin served as a loading control for total protein. (D) Wright-Giemsa staining shows the morphology of control (mock) or IL6-treated M1 cells. Scale bar, 30 μm. (E) Western blot analysis of protein lysates prepared from M1 cells expressing two different shRNAs (shAsxl1-1, shAsxl1-2) and scrambled control (Scr). β-actin served as a loading control for total protein. (F) Real-time qPCR analyses of the relative mRNA levels of Asxl1, Ink4b, Arf and p16Ink4a in control (Scr) and Asxl1-depleted (shAsxl1-1 and -2) M1 cells at different times of IL6 treatment. The error bars denote SD, n = 2. (G) Primary bone marrow cells from patients diagnosed with MDS, CMML or AML were harvested for RNA extraction followed by RT-qPCR analyses of expression levels of INK4A, INK4B and HOXA9. The bar in the middle of each group represents the median. P-values were calculated using the two-tailed Student's t-test.
In M1 myeloblastic cells, IL6 triggered cellular differentiation (Figure 3D), accompanied by an early induction of Asxl1 expression (4 h) and a strong and specific increase of Ink4b levels (Figure 3F). However, when Asxl1 was downregulated (Figure 3E and 3F), Ink4b expression was much less responsive to IL6 (Figure 3F). Similarly, we observed that the increase of Ink4b in response to LPS in M1 cells was also dependent on the expression of Asxl1 (Supplementary information, Figure S3A). Moreover, loss of Bap1 attenuates the activation of Ink4b expression induced by IL6 or LPS as efficiently as loss of Asxl1 (Supplementary information, Figure S3B and S3C).
To investigate a potential role of ASXL1 in regulating INK4B expression in patients diagnosed with MDS, CMML, or acute myeloid leukemia (AML), we compared the expression of INK4B, INK4A and HOXA9 in the primary bone marrow cells from distinct groups of patients with these diseases. These patients were clinically diagnosed with or without ASXL1 mutations (Mut or wild type, WT) as confirmed by targeted genomic DNA sequencing analysis (Supplementary information, Table S1)32. As shown in Figure 3G, the expression of INK4A is not significantly altered in Mut compared with WT. Interestingly, the expression of HOXA9 is generally higher, while the expression of INK4B is significantly lower in Mut compared with WT patients.
Taken together, our data suggest that ASXL1 has a general function of controlling the expression of INK4B in response to oncogene activation and anti-proliferative signals, and that mutations in ASXL1 lower the expression of INK4B.
ASXL1 binds to the INK4B promoter and mediates a decrease in H2AK119ub1 levels
To further understand why ASXL1 depletion specifically prevents the induction of INK4B expression, we investigated whether ASXL1 and BAP1 were bound to the INK4B locus in response to intrinsic or extrinsic signals. To avoid antibody bias, we ectopically expressed FLAG-HA-tagged Asxl1 (FH-Asxl1) in TIG3 cells at 2-3 times higher levels than the endogenous gene (Figure 4A). IP assays using anti-FLAG M2 antibody showed that the ectopic proteins can successfully reconstitute the deubiquitinase complex with BAP1 (Figure 4B). Chromatin immunoprecipitation (ChIP)-qPCR analyses using primers covering INK4B, ARF or INK4A in the FH-Asxl1 cells and parental control cells showed that FH-Asxl1 is bound to the INK4B promoter, but not at detectable levels at the promoters for ARF or INK4A (indicated as INK4B-P2 and -P3, Figure 4C). Accordingly, we observed a significant decrease of H2AK119ub1 levels specifically on the INK4B promoter in the Asxl1-overexpressing cells (Figure 4C).

ASXL1 contributes to H2A deubiquitylation at the INK4B locus. (A) Western blot analysis of total protein lysates of control and FH-Asxl1 transduced TIG3 cells. β-actin served as a loading control. (B) Ectopically expressed FH-Asxl1 interacts with BAP1, and is not associated with SUZ12 and EZH2. Anti-FLAG IP was performed in both control and FH-Asxl1 transduced TIG3 cells, followed by western blot analyses using the indicated antibodies. (C) Ectopically expressed FH-Asxl1 is enriched on the INK4B promoter, which is associated with the decreased H2AK119ub1 level at the same region. ChIP-qPCR analysis for FH-ASXL1 and H2AK119ub1 in the indicated control and FH-Asxl1 transduced TIG3 cells. Multiple primers covering INK4B-ARF-INK4A locus were examined. The error bars denote SD, n = 2. (D) ChIP-qPCR analyses on the indicated regions of the INK4B and INK4A promoters were performed for ASXL1 and BAP1, H2AK119ub1 and H3K27me3 in TIG3 ΔBRAF-ER cells expressing the indicated shRNAs and treated with or without 4-OHT. The error bars denote SD, n = 3. (E) ChIP-qPCR analyses on the Ink4b promoter were performed for Asxl1 and Bap1, H2AK119ub1 and H3K27me3 in M1 cells expressing the indicated shRNAs and treated with or without IL6. The error bars denote SD, n = 2.
ASXL1 is required for the deubiquitylation of H2AK119 at the INK4B locus
In addition to gain-of-function studies, we evaluated how ASXL1 depletion affected H2AK119ub1 levels and the regulation of the INK4B-ARF-INK4A locus in different systems. As shown in Supplementary information, Figure S4, the global levels of PcG-mediated H3K27me3 and H2AK119ub1 in control TIG3 ΔBRAF-ER cells are modestly decreased following 48 h of 4-OHT treatment, consistent with the decreased expression of PcG proteins during senescence4,30. However, when ASXL1 was depleted, the H2AK119ub1 levels remained unchanged, while the H3K27me3 levels were still decreased. These data also support a defined role for ASXL1 in regulating global levels of H2AK119ub1 in vivo.
To investigate the locus-specific effects, we first showed that endogenous ASXL1 binding is enriched at the INK4B promoter, while only a minor increase was observed at the INK4A promoter after 4-OHT treatment (Figure 4D). The specificity of the ASXL1 antibody was confirmed by the decreased enrichment on the promoters in ASXL1-depleted cells (Figure 4D). Interestingly, we noticed that BAP1 is enriched at both loci upon treatment of 4-OHT, but its binding is only affected by the loss of ASXL1 at the INK4B promoter and not at the INK4A locus (Figure 4D). Since BAP1 expression was not increased upon 4-OHT treatment, and knockdown of ASXL1 did not obviously affect the protein levels of BAP1 (Supplementary information, Figure S4), these data suggest that ASXL1 is involved in the recruitment of BAP1 to the INK4B locus rather than regulating its expression or stability. Taken together, these results demonstrate that ASXL1 is preferentially enriched at the INK4B promoter and required for BAP1 binding upon BRAF activation. In contrast, the enrichment of BAP1 on the INK4A promoter appears independent of ASXL1. It would be interesting to identify the differential regulatory mechanisms for the two closely related genes.
Next, we analyzed the local changes of PcG activity at the INK4A and INK4B locus by ChIP-qPCR. After 48 h of 4-OHT treatment, the levels of H3K27me3 and H2AK119ub1 were significantly decreased in the control cells. However, upon depletion of ASXL1, the H2AK119ub1 levels were maintained specifically at the INK4B promoter though the decrease of H3K27me3 levels were still observed at both loci (Figure 4D). These results suggest that the maintenance of H2AK119ub1 levels is sufficient for preventing the full activation of the INK4B locus by the activation of RAS-BRAF pathways (Figure 2A), and that this is independent of H3K27me3 levels.
To extend these observations to other cellular systems, we analyzed the changes of PcG activity at the Ink4b locus in M1 cells treated with IL6. ChIP-qPCR analyses showed that Bap1 enrichment at the Ink4a or Ink4b locus induced by IL6 was inhibited upon depletion of Bap1, confirming the specificity of the antibody. However, depletion of Asxl1 specifically prevented the IL6-induced Bap1 binding (Supplementary information, Figure S5A and S5B) and therefore the decrease of H2AK119ub1 levels at the Ink4b locus (Figure 4E).
Taken together, our results demonstrate that ASXL1 and BAP1 are required for the deubiquitylation of H2AK119ub1 at the INK4B locus and the increased expression of p15INK4B in response to oncogene activation and anti-proliferative signals.
ASXL1 depletion confers a context-dependent growth advantage
ASXL1 is a tumor suppressor gene and its downregulation or deletion is therefore expected to confer a growth advantage to cells. However, downregulation of ASXL1 led to slower growth of the human and mouse cells that we tested (Supplementary information, Figure S6A and S6B), suggesting that ASXL1 is also required for the activation of growth promoting genes. To test whether downregulation of ASXL1 also could confer a growth advantage, similar to what has been found in mouse genetic studies26,27,28, we tested whether the absence of ASXL1 in the presence of anti-proliferative signals would confer such a phenotype.
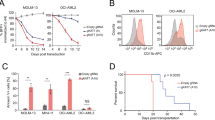
TGFβ is a potent inhibitor of epithelial cell growth that plays an important role in tissue homeostasis. Most carcinoma cells exhibit a reduced sensitivity to TGFβ-mediated growth inhibition, suggesting TGFβ participation in the development of these malignancies. It has been shown that continuous activation of TGFβ signaling for 12-14 h leads to growth arrest in HaCaT cells33. Therefore we treated the ASXL1-depleted and control HaCaT cells with TGFβ for 14 h followed by EdU incorporation-fluorescence staining assay. As shown in Figure 5A and 5B, the downregulation of ASXL1 prevented TGFβ-induced reduction in DNA replication, while the re-introduction of mAsxl1 restored the normal response to TGFβ (Figure 5C). Moreover, ASXL1-depleted HaCaT cells were also resistant to the growth inhibitory effect of TGFβ (Figure 5D and 5E).

Decreased expression of ASXL1 confers context-dependent cell growth advantages. (A) HaCaT cells expressing the indicated shRNAs were grown with or without TGFβ for 14 h, and stained for incorporation of EdU and DNA content (DAPI). The data represents one of triplicate experiments. (B, C) Quantification and statistical analysis of the fraction of EdU-positive HaCaT cells (EdU/DAPI). The absolute number of EdU-positive cells and total number of cells were counted for each group. The error bars denote SD, n = 3. (D) Colony formation assay of HaCaT cells treated as indicated. 1 000 cells of each group were seeded into one well of a six-well plate. Fresh medium was replaced every other day with or without TGFβ. After 10 days of culture, cells were washed twice with PBS and stained with crystal violet. One of the triplicate assays is shown. (E) Quantification of cell numbers of the experiment shown in D for the indicated groups of HaCaT cells. Quantifications were done as described in Materials and Methods. (F) Cell growth curves of M1 cells grown with or without IL6 and expressing the indicated shRNAs. Fresh medium was added every other day. Y-axis values represent cell numbers. The error bars denote SD, n = 2. (G) Colony formation in methylcellulose from control (Scr), ASXL1-depleted (shASXL1-B and shASXL1-C) or BAP1-depleted (shBAP1-1) primary bone marrow cells isolated from MDS patients with WT-ASXL1. CFU-GM was classified according to standard morphologic criteria. Data represent the counts of colonies from three independent experiments. Statistical significance was determined by the two-tailed Student's t-test.
To examine the effect of depleting Asxl1 or Bap1 in M1 cells, we treated the cells with IL6 for 4 days. This led to a complete growth arrest of the parental leukemogenic cells (Figure 5F), which displayed a bigger macrophage-like morphology (Figure 3D). In contrast, the Asxl1-depleted cells continued to proliferate in the presence of IL6 although at a slower rate than the untreated control M1 cells (Figure 5F, Supplementary information, Figure S6B). Bap1-depleted M1 cells grew even faster than the control cells in the absence of IL6 and did not respond to the growth inhibition of IL6 (Supplementary information, Figure S6C). Taken together, these results demonstrate that ASXL1 is required for the cellular sensitivity to extrinsic signals that induce differentiation and/or growth inhibition.
p15INK4B expression is increased during granulomonocytic and megakaryocytic differentiation, and is a regulator of myeloid differentiation34,35. Thus, we speculated that loss of ASXL1 function could confer a growth advantage to myeloid progenitors. To test this, we knocked down the expression of ASXL1 or BAP1 in primary bone marrow mononuclear cells isolated from MDS patients with WT-ASXL1, and compared their growth potential with control cells in a colony-forming unit-granulocyte-macrophage (CFU-GM) assay. With equal number of cells seeded, significantly more colonies were formed from the ASXL1- or BAP1-depleted samples comparing with the control sample (Figure 5G). Hence the in vitro data demonstrate that the loss of ASXL1 or BAP1 confers a higher proliferative capacity to the myeloid progenitor cells, similar to the phenotype observed in Ink4b−/− in mice34.
Discussion
In this study, we have demonstrated that ASXL1 is required for the activation of INK4B locus in response to oncogenic and anti-proliferative signaling, through a mechanism that most likely involves the removal of the PcG repressive modification H2AK119ub1 (Figure 6).

Model for the role of ASXL1 in the regulation of the INK4B locus. Oncogene induction and anti-proliferative signals lead to the recruitment of ASXL1 and BAP1 to the INK4B locus. This recruitment leads to the deubiquitylation of H2AK119ub1 associated with the locus and the increased expression of INK4B. The increased expression leads subsequently to cell growth arrest or differentiation. In contrast, when ASXL1 is mutated, cells are resistant to the anti-proliferative signals, which correlates with the failure to deubiquitylate H2AK119ub1 and increase the expression of p15INK4B. Since we have only shown that OGT, HCF-1, and FOXK1,2 are in a complex with ASXL1 and BAP1 by mass spectrometry analysis and not that the proteins bind to the INK4B locus, this part of the model is speculative.
ASXL1 mutations were reported in diverse malignancies and it was recently confirmed to have a tumor suppressor activity in hematopoietic cells26,27,28,29. In addition, BAP1 mutations are frequently found in mesotheliomas (21%-23%)36,37, melanomas (47% of Uveal Melanoma, more than 80% of metastasizing uveal melanoma38,39) and sporadically some other tumors (breast, bladder, renal, lung, ovary, and pancreas). Deletion of BAP1 in hematopoietic lineages also leads to MDS-like abnormalities40 and confers greater proliferation potential to the myeloid progenitors (Figure 5G), suggesting that ASXL1 and BAP1 have overlapping functions in the hematopoietic system. In line with the functional overlap, our results demonstrate that ASXL1 is essential for the deubiquitylation activity of BAP1 and for the transcriptional activation of INK4B.
Recently, it was demonstrated that ASXL1 directly interacts with PRC2, and that ASXL1 ablation leads to PRC2 dissociation from chromatin and therefore loss of H3K27me3 activity28,29. In our studies, we were not able to confirm a physical interaction between ASXL1 and PRC2 (Figure 1, Supplementary information, Figure S1 and Figure 4A). In addition, loss of ASXL1 did not lead to any detectable changes in H3K27me3 levels, and the role of ASXL1 in regulating the INK4B locus therefore appears to be independent of a direct interaction with PRC2. Instead, we found that HCF1, a common partner for ASXL1 and BAP1, interacts with several proteins involved in transcriptional activation such as WDR5, ASH2L, SET1A, and KMT2A (Supplementary information, Figure S1C). Although our results do not exclude a direct or indirect role of ASXL1 in the repression of oncogenes such as HOXA9, the demonstration that ASXL1 binds directly to the INK4B locus and is required for its activation in response to oncogene activation provides striking support for the potential mechanism by which ASXL1 restricts cell proliferation.
p15INK4B belongs to a family of four cyclin-dependent kinase inhibitors that form a complex with CDK4 or CDK6 to inhibit their kinase activity. The silencing of the INK4B locus is known to provide the ability of tumor cells to escape the G1 checkpoint of the cell cycle, which thereby can escape cell cycle arrest11. INK4B is particularly important in the hematopoietic system, where it is induced by a plethora of cytokines and infectious agents, and thus plays an important role in the regulation and control of hematopoiesis. Conditional myeloid-lineage deletion of Ink4b in mice results in an expansion of myeloid and monocytic cells in the bone marrow, and leads to the development of myeloproliferative neoplasms (MPN)-like disorders34. Interestingly, the downregulation of ASXL1 provides a growth advantage to hematopoietic progenitors, conferring impaired myeloid differentiation (Figure 5F and 5G,27) and poor prognosis in either MDS23 or CMML41 patients. Moreover, the INK4B promoter in the INK4B-ARF-INK4A locus is exclusively methylated in 50%-60% of patients with MDS and 70%-80% patients with AMLs42. Silencing of INK4B locus by DNA methylation was also found to contribute to the disease evolution from MDS to AML43,44 or acute lymphoblastic leukemia44. Currently, there are no data sets available that have analyzed the co-occurrence of ASXl1 mutations and INK4B methylation. However, several results support the notion that gene silencing precedes DNA methylation45,46. Thus, it is tempting to speculate that mutations of ASXL1 lead to DNA methylation of the INK4B promoter in hematological diseases. Based on these data, we propose that therapeutic strategies for cancer patients with ASXL1 mutations should consider de-repressing epigenetically silenced INK4B47 by the use of DNA methylation inhibitors, which are already in use for MDS patients.
Materials and Methods
Cloning and plasmid preparation
Human ASXL1 cDNA was cloned from human fetal cDNA library. Mouse cDNA of Asxl1 was a gift from Qun-Tian Wang lab in University of Chicago. They were introduced into Gateway Entry vector pCR8/GW/TOPO (Invitrogen) following the manufacturer's protocol and verified by sequencing. Different constructs were subcloned into the desired vectors by Gateway technology (Invitrogen).
Cell culture
Flp-In T-REX 293 cells (Life technologies), human diploid lung embryonic fibroblast cell line TIG3, human keratinocytes HaCaT were grown in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS). 1 mM 4-OHT was added to the culture medium to activate ΔBRAF in TIG3 ΔBRAF-ER cells. The murine myeloid cell line M1 (ATCC) and the human AML cells line NOMO1 (DMZ ACC542) were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS. For induction of growth arrest, HaCaT cells were cultured with 2 ng/ml of TGFβ1 (R&D, referred to as TGFβ throughout manuscript). M1 cells were seeded at a concentration of 105/ml and continuously cultivated in medium containing 50 ng/mL of IL6 (R&D). The Flp-In T-REx 293 cell lines with stable inducible expression of FH-ASXL1, FH-BAP1 and HCF1 proteins in response to the tetracycline were generated following the manufacturer's instructions. Briefly, the parental Flp-In T-REx 293 cells were co-transfected with pcDNA5/FRT-FLAG-HA-ASXL1, pcDNA5/FRT-FLAG-HA-BAP1 or pcDNA5/FRT-FLAG-HA-HCF1 and the Flp recombinase vector pOG44. After hygromycin selection, positive clones were validated by western blot. The cells were then expanded for further experiments.
EdU staining and colony formation assay
EdU staining was performed using the Click-iT EdU Alexa Fluor 488 Imaging Kit (Life Technologies) according to the provided protocols. For colony formation assays using HaCaT cells, 1 000 cells were seeded onto one well of 6-well plates for each group. After 10 days the colonies were stained using crystal violet. The assay was performed in triplicate and one six-well plate for each condition is shown. To quantify the differences of cell numbers, we de-stained the crystal violet with 10% acetic acid after staining for 15 min. Then the samples from each group were diluted with water and their absorbance was measured at 590 nm with spectrophotometer.
Primary patient samples and ASXL1 genomic DNA sequencing analysis
Approval was obtained from the Ethics Committees of the Institute of Hematology, Chinese Academy of Medical Sciences and Peking Union Medical College and informed consent was provided according to guidelines of the Declaration of Helsinki. The genomic DNA sequence analysis for ASXL1 was performed as described32.
Colony-forming unit granulocyte macrophage assay
Primary bone marrow cells isolated from MDS patients containing wild-type ASXL1 were transduced with a lentivirus expressing shRNAs against ASXL1 or BAP1. After 3 days of selection, 105 bone marrow cells were cultured in semisolid Metho-Cult H4434 Classic methylcellulose (StemCell Technologies, Manchester, UK) according to the manufacturer's instructions. Assessment of the colony numbers was performed after 14 days in culture. Experiments were performed in triplicate.
RNA interference with short hairpin RNAs and genome editing with CRISPR/CAS9 system
Specific oligonucleotides against human ASXL1 (shASXL1-B: GCTGATGGTGAATTTACTCAT; shASXL1-C: GCTTCTGTAAGTATGCTCTAT) and mouse Asxl1 (shAsxl1-1: GTGGAAAGCTGTGGGTCTA, shAsxl1-2: GCTGATGGTGAATTCACTCAT) were designed and cloned into pLKO.1 according to the protocol recommended by Addgene. If not specified, shASXL1-B or shAsxl1-2 was used in human or mouse cells. The shRNA constructs against human or mouse BAP1 in the LKO.1-puro vector were from Sigma (shBAP1-1: TRCN0000007370, shBAP1-2: TRCN0000007372 and shBap1-1: TRCN0000030719, shBap1-2: TRCN0000030723). Two sgRNAs for mouse Asxl1 (GTGAAAAGACTAATGCGGCC) and Bap1 (GGATCGAAGAGCGCAGGTCC) were designed and cloned into the vector lentiCRISPR v248 (lentiCRISPR v2 was a gift from Feng Zhang, Addgene plasmid # 52961).
Lentivirus preparation
All lentiviruses were generated as follows: Superfect-mediated co-transfection of lentiviral backbone with pAX8 (packaging) and pCMV-VSVG (envelope) into 293FT cells. After 48 h, virus supernatants were column-concentrated and incubated with TIG3 or HaCaT cell suspension along with polybrene (8 μg/ml; Sigma). When appropriate, Neomycin (0.3 mg/ml) and/or Puromycin-containing media (2 μg/ml) was replaced 48 h postinfection for an additional 96 h before testing the expression.
Generation of antibodies
The polyclonal antibodies to Asxl1 were produced in rabbits using synthetic peptides corresponding to amino acids 858-869 (LLIRESSRQEAL) and 881-896 (WVPILSNYEVIKTSDP) of mouse Asxl1, respectively. These peptides were coupled to Keyhole Limpet Hemocyanin (KLH) through the C- or N-terminal lysine residues, respectively, and injected subcutaneously into rabbits. Specific antibodies from positive sera were affinity-purified on the respective peptide antigens according to standard procedures. Mouse polyclonal and monoclonal antibodies for ASXL1 and mouse monoclonal antibodies for BAP1 were generated using standard immunization procedures and insect cell-expressed full-length recombinant human ASXL1 or BAP1 as antigens.
Size-exclusion chromatography
Nuclear extracts from 293 cells were prepared as described49, lysed in high salt buffer (50 mM Tris-HCl, pH 7.6, 300 mM NaCl, 10% glycerol, 0.2% (v/v) Igepal CA 630) and fractionated on a Superose 6 PC 3.2/30 (GE Healthcare) on an AKTA purifier system (GE Healthcare). Fractions 12-36 were loaded for western blot analyses.
In vitro deubiquitylation assays
Full-length recombinant ASXL1, BAP1 WT and catalytically inactive mutant (A95D) were purified by FLAG-affinity chromatography from insect cells expressing FLAG-histidine tag versions of the proteins as described50. Nucleosomes purified from HeLa cells51 were incubated with the specified recombinant proteins at room temperature for 1 h in deubiquitylation buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM MgCl2, 1 mM ZnCl2, 1 mM dithiothreitol (DTT)). The reactions were stopped by the addition of equal volume of 2× SDS sample loading buffer and boiled for 5 min.
Real-time qPCR, immunoblotting and immunostaining
Total RNA was extracted with the RNeasy Mini Kit (Qiagen) and reverse transcribed using the TaqMan Reverse Transcription Reagents kit (Applied Biosystems). RPLPO was used as a reference gene for all quantitative RT-PCR experiments and analyses. To obtain whole-cell protein extracts, cells were lysed in RIPA buffer. Acid extraction of histones was performed according to the Abcam protocol52. The immunostaining of p15INK4B was carried out as described53. Primer sequences and information for antibodies are available in Supplementary information, Table S2.
Complex purification and identification, IP, and ChIP
The FLAG-HA tandem affinity purification was performed followed by MS analysis. Nuclear extracts (200 mg, 2 × 109 cells) from the Flp-In T-REx 293 cells stably expressing a FLAG-HA-tagged protein was incubated with a 300 μl packed volume of anti-Flag-beads (M2-agarose, Sigma) overnight at 4 °C with rotation. The beads were collected by centrifugation at 700× g for 5 min and washed 6 times with 40× resin bed volume of buffer A (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 10% glycerol, 0.2 mM PMSF, 1 mM DTT, 1 μg/ml aprotinin and 1 μg/ml leupeptin). The beads were transferred into a 10 ml poly-prep chromatography column (Bio-Rad) and complexes were then eluted five times after 10 min of incubation using one resin bed volume of buffer A supplemented with 0.5 μg/μl FLAG peptide. The FLAG-IP elute was incubated with 100 μl of a 50 % slurry of HA-beads overnight. The beads were washed four times with buffer A and eluted with 100 μl buffer A supplemented with 1 μg/μl HA peptide for 2 h. The samples were boiled in SDS loading buffer and run shortly on SDS-PAGE gel. A gel slice containing the purified proteins was isolated for MS analysis. Briefly, the MS analysis was performed by using a standard proteomics workflow. The proteins were in-gel digested using 10 mM DTT (45 min at 56 °C), followed by 40 mM iodoacetamide (30 min at room temperature in dark) and overnight incubation with trypsin (enzyme:sample ratio ∼1:20, pH 8.0, 37 °C). Separation and MS detection were achieved by using nano liquid chromatography coupled online to tandem MS using a data dependent acquisition method.
Regular IPs were performed in IP (250) buffer (50 mM Tris-HCl, pH 7.5, 250 mM NaCl, 5% glycerol, 0.2% Igepal, Aprotenin, Leupeptin, 0.5 PMSF, 0.5 mM DTT). Conventional and H2AK119ub1 ChIPs were performed as described52. Primer sequences for amplification of genomic DNA are available in Supplementary information, Table S2.
Statistics
Significant differences for all quantitative data were considered when P < 0.05; *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
References
Kim WY, Sharpless NE . The regulation of INK4/ARF in cancer and aging. Cell 2006; 127:265–275.
Sherr CJ . The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2001; 2:731–737.
Aguilo F, Zhou MM, Walsh MJ . Long noncoding RNA, polycomb, and the ghosts haunting INK4b-ARF-INK4a expression. Cancer Res 2011; 71:5365–5369.
Bracken AP, Kleine-Kohlbrecher D, Dietrich N, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 2007; 21:525–530.
Muller J, Verrijzer P . Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr Opin Genet Dev 2009; 19:150–158.
Simon JA, Kingston RE . Mechanisms of Polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 2009; 10:697–708.
Popov N, Gil J . Epigenetic regulation of the INK4b-ARF-INK4a locus: In sickness and in health. Epigenetics 2010; 5:685–690.
Gil J, Peters G . Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 2006; 7:667–677.
Peters G . An INKlination for epigenetic control of senescence. Nat Struct Mol Biol 2008; 15:1133–1134.
Amanullah A, Hoffman B, Liebermann DA . Deregulated E2F-1 blocks terminal differentiation and loss of leukemogenicity of M1 myeloblastic leukemia cells without abrogating induction of p15(INK4B) and p16(INK4A). Blood 2000; 96:475–482.
Hannon GJ, Beach D . p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 1994; 371:257–261.
Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J . TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol 2001; 3:400–408.
Krimpenfort P, Ijpenberg A, Song JY, et al. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 2007; 448:943–946.
Sinclair DA, Milne TA, Hodgson JW, et al. The Additional sex combs gene of Drosophila encodes a chromatin protein that binds to shared and unique Polycomb group sites on polytene chromosomes. Development 1998; 125:1207–1216.
Scheuermann JC, de Ayala Alonso AG, Oktaba K, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010; 465:243–247.
Fisher CL, Lee I, Bloyer S, et al. Additional sex combs-like 1 belongs to the enhancer of trithorax and Polycomb Group and genetically interacts with Cbx2 in mice. Dev Biol 2009; 337:9–15.
Boultwood J, Perry J, Pellagatti A, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia 2010; 24:1062–1065.
Carbuccia N, Murati A, Trouplin V, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009; 23:2183–2186.
Carbuccia N, Trouplin V, Gelsi-Boyer V, et al. Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias. Leukemia 2010; 24:469–473.
Roche-Lestienne C, Marceau A, Labis E, et al. Mutation analysis of TET2, IDH1, IDH2 and ASXL1 in chronic myeloid leukemia. Leukemia 2011; 25:1661–1664.
Stephens PJ, Tarpey PS, Davies H, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012; 486:400–404.
Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012; 487:239–243.
Thol F, Friesen I, Damm F, et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol 2011; 29:2499–2506.
Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011; 364:2496–2506.
Gelsi-Boyer V, Trouplin V, Roquain J, et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol 2010; 151:365–375.
Wang J, Li Z, He Y, et al. Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood 2014; 123:541–553.
Inoue D, Kitaura J, Togami K, et al. Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. J Clin Invest 2013; 123:4627–4640.
Abdel-Wahab O, Gao J, Adli M, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med 2013; 210:2641–2659.
Abdel-Wahab O, Adli M, Lafave LM, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012; 22:180–193.
Agger K, Cloos PA, Rudkjaer L, et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev 2009; 23:1171–1176.
Schmidt M, Bies J, Tamura T, Ozato K, Wolff L . The interferon regulatory factor ICSBP/IRF-8 in combination with PU.1 up-regulates expression of tumor suppressor p15(Ink4b) in murine myeloid cells. Blood 2004; 103:4142–4149.
Wang J, Ai X, Gale RP, et al. TET2, ASXL1 and EZH2 mutations in Chinese with myelodysplastic syndromes. Leuk Res 2013; 37:305–311.
Nicolas FJ, Hill CS . Attenuation of the TGF-beta-Smad signaling pathway in pancreatic tumor cells confers resistance to TGF-beta-induced growth arrest. Oncogene 2003; 22:3698–3711.
Rosu-Myles M, Taylor BJ, Wolff L . Loss of the tumor suppressor p15Ink4b enhances myeloid progenitor formation from common myeloid progenitors. Exp Hematol 2007; 35:394–406.
Teofili L, Morosetti R, Martini M, et al. Expression of cyclin-dependent kinase inhibitor p15(INK4B) during normal and leukemic myeloid differentiation. Exp Hematol 2000; 28:519–526.
Bott M, Brevet M, Taylor BS, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet 2011; 43:668–672.
Testa JR, Cheung M, Pei J, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 2011; 43:1022–1025.
Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 2011; 43:1018–1021.
Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010; 330:1410–1413.
Dey A, Seshasayee D, Noubade R, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 2012; 337:1541–1546.
Itzykson R, Kosmider O, Renneville A, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol 2013; 31:2428–2436.
Drexler HG . Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia 1998; 12:845–859.
Quesnel B, Guillerm G, Vereecque R, et al. Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood 1998; 91:2985–2990.
Herman JG, Civin CI, Issa JP, Collector MI, Sharkis SJ, Baylin SB . Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res 1997; 57:837–841.
Stirzaker C, Song JZ, Davidson B, Clark SJ . Transcriptional gene silencing promotes DNA hypermethylation through a sequential change in chromatin modifications in cancer cells. Cancer Res 2004; 64:3871–3877.
Schubeler D . Function and information content of DNA methylation. Nature 2015; 517:321–326.
Geyer CR . Strategies to re-express epigenetically silenced p15(INK4b) and p21(WAF1) genes in acute myeloid leukemia. Epigenetics 2010; 5:696–703.
Sanjana NE, Shalem O, Zhang F . Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014; 11:783–784.
Pasini D, Hansen KH, Christensen J, Agger K, Cloos PA, Helin K . Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev 2008; 22:1345–1355.
Christensen J, Agger K, Cloos PA, et al. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 2007; 128:1063–1076.
Wu X, Gong Y, Yue J, Qiang B, Yuan J, Peng X . Cooperation between EZH2, NSPc1-mediated histone H2A ubiquitination and Dnmt1 in HOX gene silencing. Nucleic Acids Res 2008; 36:3590–3599.
Wu X, Johansen JV, Helin K . Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol Cell 2013; 49:1134–1146.
Cloos PA, Christensen J, Agger K, et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006; 442:307–311.
Acknowledgements
We thank members of the Helin laboratory for discussions, technical advice and support. The work in the Helin lab was supported by grants from the Excellence Program of the University of Copenhagen, the Danish Cancer Society, the Novo Nordisk Foundation, and the European Commission's 7th Framework Program 4DCellFate (277899). The work in the Helin and the Jensen labs was supported by the Danish National Research Foundation (DNRF82). The work in the Rappsilber lab was supported by Wellcome Trust (103139, 092076, and 091020). The Project is sponsored by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry. The work also received support from the National Natural Science Foundation of China (81470295 to ZX, 81171899 to Xi Wang, 31570774 to Xudong Wu and 81170007 to GX).
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
ASXL1 and BAP1 form a complex similar to PR-DUB, separate from PRC2. (PDF 166 kb)
Supplementary information, Figure S2
Depletion of ASXL1 attenuates TGFβ-induced p15INK4B expression. (PDF 283 kb)
Supplementary information, Figure S3
Downregulation of Asxl1 or Bap1 attenuates IL6- and LPS-induced p15Ink4b activation in M1 cells. (PDF 239 kb)
Supplementary information, Figure S4
ASXL1 is required for oncogenic BRAF-induced decrease of H2AK119ub1 levels. (PDF 250 kb)
Supplementary information, Figure S5
IL6-induced Bap1 binding is prevented by the depletion of Asxl1 specifically at the Ink4b locus. (PDF 142 kb)
Supplementary information, Figure S6
Depletion of ASXL1 inhibits cell growth. (PDF 225 kb)
Rights and permissions
About this article

Cite this article
Wu, X., Bekker-Jensen, I., Christensen, J. et al. Tumor suppressor ASXL1 is essential for the activation of INK4B expression in response to oncogene activity and anti-proliferative signals. Cell Res 25, 1205–1218 (2015). https://doi.org/10.1038/cr.2015.121
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2015.121
Keywords
This article is cited by
-
BAP1 in cancer: epigenetic stability and genome integrity
Discover Oncology (2022)
-
The integrated stress response is tumorigenic and constitutes a therapeutic liability in KRAS-driven lung cancer
Nature Communications (2021)
-
Perithyroidal Salivary Gland Acinic Cell Carcinoma: Morphological and Molecular Attributes of a Unique Lesion
Head and Neck Pathology (2021)
-
BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation
Nature Communications (2019)
-
The role of ASXL1 in hematopoiesis and myeloid malignancies
Cellular and Molecular Life Sciences (2019)
