For more than a year now, scientists and clinicians have been trying to understand why some people develop severe COVID-19 whereas others barely show any symptoms. Risk factors such as age and underlying medical conditions1, and environmental factors including socio-economic determinants of health2, are known to have roles in determining disease severity. However, variations in the human genome are a less-investigated source of variability. Writing in Nature, members of the COVID-19 Host Genetics Initiative3 (www.covid19hg.org) report results of a large human genetic study of SARS-CoV-2 infection. The researchers identify 13 locations (or loci) in the human genome that affect COVID-19 susceptibility and severity.
Read the paper: Mapping the human genetic architecture of COVID-19
Scientists already knew that human genetic variants can influence the severity of infectious diseases, including infection with SARS-CoV-24-6. The effects of genetic factors range from those of rare, high-impact mutations that can make the difference between an individual developing mild symptoms and life-threatening illness7, to more-common genetic variants that only moderately affect symptom severity5.
Even so, human genomic studies of infectious diseases remain scarce compared with those of other immune-mediated conditions, such as autoimmune disorders. There are several reasons for that. Chief among them is that infectious diseases are typically studied with a focus on the disease-causing microorganism, rather than the host. Moreover, human genetic variants usually have relatively small effects on infection outcomes compared with the effects of socio-demographic factors such as age or access to health care8. Identifying these generally modest effects requires studies of large, well-characterized groups of people to produce sufficient statistical power to reveal the relevant genetic factors. Finally, unlike for chronic diseases, the window for characterizing the severity and outcomes of infectious diseases is often limited to a short period during which individuals are symptomatic.
The authors overcame these challenges by rapidly setting up a large, international collaboration when the pandemic started. This collaboration of around 3,000 researchers and clinicians includes data from 46 studies involving more than 49,000 individuals with COVID-19 and 2 million control individuals, with participants recruited from 6 ancestry groups and 19 countries. By acting swiftly, the authors could recruit symptomatic patients, and, by setting up international collaborations, were able to include enough participants to overcome statistical-power limitations. In addition, they tried to account for the role of socio-demographic factors by collecting data on some of the known risk factors, such as age and sex, and including this information in their statistical analyses.
To obtain comparable results across all 46 study groups, the authors defined 3 categories of analysis: infection, which included people with physician-confirmed, laboratory-confirmed or self-reported COVID-19; hospitalization, which consisted of individuals with laboratory-confirmed moderate to severe COVID-19; and critical illness, patients with laboratory-confirmed infection who were hospitalized and required respiratory support or died. To identify genetic variants associated with COVID-19 susceptibility and severity, the authors first compared the difference in the frequencies of millions of genetic variants between the people infected with COVID-19 and the control individuals in each study. They then combined the results from all 46 studies to increase the statistical power of their data.
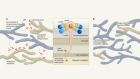
Through this combined analysis, the authors identified 13 loci that were associated with SARS-CoV-2 infection and disease severity (Fig. 1), including 6 loci not reported in previous human genomics studies of COVID-194,5. Four loci affect general susceptibility to SARS-CoV-2, whereas nine were associated with disease severity. Two of the previously unassociated loci were discovered only when individuals with East Asian ancestry were included in the analysis, highlighting the value of including diverse populations in human genomics studies.

Figure 1 | Identifying regions of the human genome associated with COVID-19 susceptibility and severity. The COVID-19 Host Genetics Initiative3 sought to identify genetic variants that account for the variability in individuals’ susceptibility to COVID-19, as well as in the severity of the disease. The authors compared the genomes of 49,562 individuals with COVID-19 (including 13,641 individuals who were hospitalized with the infection and, of those, 6,179 who were critically ill with the disease) with the genomes of around 2 million control individuals without known infection. This comparison pointed to 13 locations in the genome (loci): variants in 4 of these loci are associated with susceptibility to COVID-19, whereas variants in 9 others are associated with disease severity.
To better understand the biology of COVID-19 and the mechanisms that connect these loci to disease outcomes, the authors looked for genes that were in the proximity of each locus (that is, ‘candidate genes’). They identified more than 40 candidate genes, several of which have previously been implicated in immune function or have known functions in the lungs, suggesting that variants in the genomic regions highlighted by the authors’ findings might exert their effect on COVID-19 outcome through the respiratory system.
One such example is the gene TYK2. Variants of this gene can increase susceptibility to infections by other viruses, bacteria and fungi9. In line with this, the authors reported that individuals who carry certain mutations in TYK2 are at increased risk of being hospitalized or developing critical illness from infection with SARS-CoV-2. Another example is the gene DPP9. The authors found a variant in this gene that increases the risk of becoming critically ill with COVID-19. Notably, the same variant can increase the risk of a rare pulmonary disease characterized by scarring of the lung tissue10.
A race to determine what drives COVID-19 severity
This study by the COVID-19 Host Genetics Initiative represents a major milestone in our understanding of the role of human genetics in susceptibility to SARS-CoV-2; however, more work remains to be done. Future experiments should determine all the genes, signalling pathways and biological mechanisms that connect the genomic loci identified to COVID-19 outcomes.
Moreover, despite the authors’ efforts to include genetically diverse study groups, about 80% of the participants are of European ancestry. Future studies containing a larger number of individuals from other ancestry groups are needed to ensure that the results apply to non-Europeans, and to identify other loci that might be associated with risk in people of other ancestries.
Another complex question that could not be addressed in the authors’ study is the combined effect of specific variants in the SARS-CoV-2 genome and variants in the human genome on disease outcome. Finally, as the authors mention, they could not fully control for all socio-demographic factors, such as access to health care. Although such non-genetic factors are unlikely to explain all the findings, they could bias some of the associations between genetic variants and disease outcome.
Despite these limitations, the implications of the study’s results are far-reaching. This study is important not only for advancing our understanding of human susceptibility to COVID-19; it also underlines the value of global collaborations for clarifying the human genetic basis of variability in susceptibility to infectious diseases. Infections remain among the top causes of mortality in lower-income countries, and represent a growing global threat, owing to climate change, urbanization and rising population size11. Human genomics can be an effective tool with which to understand the biological mechanisms that underlie immune responses to specific infections, to identify at-risk individuals and to develop new drugs and vaccines for existing or emerging infections.
 Read the paper: Mapping the human genetic architecture of COVID-19
Read the paper: Mapping the human genetic architecture of COVID-19
 A race to determine what drives COVID-19 severity
A race to determine what drives COVID-19 severity
 Neanderthal DNA highlights complexity of COVID risk factors
Neanderthal DNA highlights complexity of COVID risk factors