- NEWS AND VIEWS
Activation of retinal neurons triggers tumour formation in cancer-prone mice
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 594, 179-180 (2021)
doi: https://doi.org/10.1038/d41586-021-01353-9
References
Pan, Y. et al. Nature 594, 277–282 (2021).
Bajenaru, M. L. et al. Cancer Res. 63, 8573–8577 (2003).
Toonen, J. A., Ma, Y. & Gutmann, D. H. Neuro Oncol. 19, 808–819 (2017).
Venkatesh, H. S. et al. Cell 161, 803–816 (2015).
Venkataramani, V. et al. Nature 573, 532–538 (2019).
Venkatesh, H. S. et al. Nature 573, 539–545 (2019).
Venkatesh, H. S. et al. Nature 549, 533–537 (2017).
Magnon, C. et al. Science 341, 1236361 (2013).
Mauffrey, P. et al. Nature 569, 672–678 (2019).
Saloman, J. L. et al. Proc. Natl Acad. Sci. USA 113, 3078–3083 (2016).
Peterson, S. C. et al. Cell Stem Cell 16, 400–412 (2015).
Monje, M. et al. Cell 181, 219–222 (2020).
Competing Interests
The authors declare no competing interests.
 Read the paper: NF1 mutation drives neuronal activity-dependent initiation of optic glioma
Read the paper: NF1 mutation drives neuronal activity-dependent initiation of optic glioma
 Brain tumours manipulate neighbouring synapses
Brain tumours manipulate neighbouring synapses
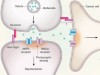 Dangerous liaisons as tumour cells form synapses with neurons
Dangerous liaisons as tumour cells form synapses with neurons