The most life-threatening complication of COVID-19 is severe pneumonia with intense damage to the air sacs of the lungs. Inflammation has a central role in this process. Although inflammation is an essential part of our immune defences to control infection, it can nevertheless cause collateral damage to tissues. Because of the high death rate and long duration of illness observed with COVID-19 pneumonia, it has been suggested that the inflammation that arises from infection with SARS-CoV-2 (the coronavirus that causes COVID-19) is somehow different from the type of inflammation caused by other respiratory infections. However, there was a lack of comprehensive data to support this claim. Writing in Nature, Grant et al.1 address this knowledge gap by directly comparing immune cells isolated from the lungs of people with COVID-19 pneumonia with immune cells obtained from people who had pneumonia that arose from other causes.
Read the paper: Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia
Like most viruses that infect the respiratory system, SARS-CoV-2 can enter the body through the nose or mouth, and infect the epithelial cells that line the surface of the respiratory tract in the throat and lungs. Infected cells release molecular signals that alert immune cells in the blood to migrate to the lung to augment antiviral responses2. Although important information can be gleaned by studying such immune cells found in the bloodstream, a true understanding of lung inflammation requires research that involves the isolation of these cells from the air sacs (alveoli) of the lungs.
The gold-standard approach to obtain immune cells from the lungs is a technique termed bronchoalveolar lavage, in which saline is introduced into the small airways and air sacs and then gently removed using suction to retrieve the cells of interest. Using this method, Grant and colleagues obtained immune cells (Fig. 1) from 88 people who had COVID-19 pneumonia and 211 people who had pneumonia that arose from other infections. All samples came from people in intensive-care units who required a mechanical ventilator to breathe. In most cases, samples were obtained at repeated intervals during illness, providing a comprehensive analysis over time (a longitudinal analysis) of the inflammatory response in the lungs of people with severe COVID-19 pneumonia.

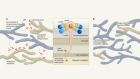
Figure 1 | A cycle of inflammation that might recruit immune cells to the lungs during COVID-19 pneumonia. Grant et al.1 analysed immune cells from the lungs of people who had pneumonia arising from COVID-19 or other conditions. Individuals with COVID-19 had a greater proportion of T cells and monocytes in their immune-cell population than did individuals with other types of pneumonia. a, The authors confirmed reports4,5 that, in the lungs, immune cells called macrophages are infected by the SARS-CoV-2 virus that causes COVID-19. Grant et al. found that these infected cells express genes encoding the types of chemoattractant molecule that can recruit T cells and monocytes to the lungs from blood. b, T cells that entered the lungs expressed the gene encoding the inflammatory molecule interferon-γ (IFN-γ). IFN-γ might drive monocytes and macrophages to boost production of chemoattractants and other inflammatory molecules (not shown). c, Monocytes could mature into macrophages and restart the cycle if infected with SARS-CoV-2. The proposed loop of inflammation and immune-cell recruitment could explain the lengthy inflammation that characterizes COVID-19 pneumonia.
In the first step of their study, the authors analysed the different classes of inflammatory cell isolated from the air sacs. Intriguingly, the authors found that the immune cells from people who had COVID-19 contained a greater proportion of T cells and monocytes, compared with the immune-cell population of people whose pneumonia arose from a different cause. T cells and monocytes are relatively rare in healthy lungs3, so the assumption is that these immune cells were recruited to the air sacs by locally produced chemoattractant proteins. Importantly, these differences in inflammatory cell types in people with COVID-19 could not be explained by other clinical factors, such as the phase of their illness or a co-infection with other disease-causing agents.
To investigate the mechanisms underlying the recruitment of T cells and monocytes to the lungs, Grant et al. tested whether immune cells were infected with SARS-CoV-2. They confirmed previous reports4,5 that RNA of this virus is found in macrophage cells — a type of immune cell known to take up (engulf) dead cells and debris — in the lungs. Interestingly, the authors detected viral RNA corresponding to both the ‘positive’ strand of the virus, which encodes viral proteins, and the mirror-image copy of this strand (the ‘negative’ strand). The presence of the negative strand is particularly suggestive of active replication of the virus in macrophages. These macrophages also had high levels of expression of genes encoding chemoattractant proteins such as CCL4 and CXCL10 that can recruit T cells and monocytes. The authors therefore reasoned that macrophage infection by SARS-CoV-2 triggers the infected cells to release these chemoattractants.
Interferon deficiency can lead to severe COVID
To determine possible roles for T cells that are recruited to the lungs, the authors assessed gene expression at the level of single cells. Grant and colleagues found that the T cells expressed RNA encoding the protein interferon-γ (IFN-γ), which is known to stimulate macrophages and monocytes to produce inflammatory molecules6. Indeed, the authors found that macrophages from people with COVID-19 expressed interferon-responsive genes at higher levels than did macrophages from people who didn’t have COVID-19.
One of the unique characteristics of severe COVID-19 pneumonia is its particularly long duration, compared with the typical duration of pneumonia associated with other viral infections. Severe lung inflammation and prolonged respiratory failure persist in COVID-19 even after the viral infection is no longer detectable7. However, the mechanisms that drive this sustained inflammatory response have not been fully determined. One possibility is a positive feedback loop, as proposed by Grant and colleagues, in which macrophage infection by SARS-CoV-2 leads to the production of chemoattractants that recruit monocytes and T cells to the lungs (Fig. 1). The newly recruited monocytes might mature to form macrophages, and if the T cells produce IFN-γ that stimulates macrophages to produce more chemoattractants, this would perpetuate a cycle of inflammation.
COVID-19 poses a riddle for the immune system
Although the data supporting the authors’ model are based almost exclusively on gene-expression profiling of immune cells, this scheme offers a unifying and plausible explanatory framework. To prove that this model is correct, each proposed step would need to be validated in functional studies, and the cause-and-effect relationship of each suggested link in this pathway should be tested. In addition, careful consideration should be paid to how the function of these immune cells of interest might integrate with the roles of other types of lung cell, such as epithelial cells, which are the main type of cell infected by SARS-CoV-2. Epithelial cells have been implicated previously4 as being the primary driver of inflammation in COVID-19.
It is notable that the types of cell and molecule that comprise this proposed self-sustaining inflammatory circuit in COVID-19 are also present to some degree8 in cells isolated from people with pneumonia arising from other types of bacterial or viral infection. This begs the question of whether the inflammatory pathway uncovered by the authors is specific to COVID-19, or whether it also operates in other forms of severe pneumonia. Confirmation of either possibility would represent a major advance. This is because such a discovery might lead to therapeutic targeting of infected macrophages, inflammatory T cells or specific inflammatory molecules as a way to block self-sustaining inflammatory circuits, and thereby offer a way to prevent persistent lung injury.
 Read the paper: Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia
Read the paper: Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia
 COVID-19 poses a riddle for the immune system
COVID-19 poses a riddle for the immune system
 Interferon deficiency can lead to severe COVID
Interferon deficiency can lead to severe COVID