- NEWS FEATURE
- Clarification 10 November 2020
Are infections seeding some cases of Alzheimer’s disease?

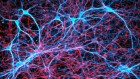
Some scientists think that microbes such as the herpes simplex virus 1 (shown here on an epithelial cell) could trigger some cases of Alzheimer’s disease. Credit: SPL
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 587, 22-25 (2020)
doi: https://doi.org/10.1038/d41586-020-03084-9
Updates & Corrections
-
Clarification 10 November 2020: An earlier version of this story did not give the primary affiliation for Yue-Ming Li.
References
Jamieson, G. A., Maitland, N. J., Wilcock, G. K., Craske, J. & Itzhaki, R. F. J. Med. Virol. 33, 224–227 (1991).
Readhead, B. et al. Neuron 99, 64–82 (2018).
Allnutt, M. A. et al. Neuron 105, 1027–1035 (2020).
Tzeng, N. S. et al. Neurotherapeutics 15, 417–429 (2018).
Soscia, S. J. et al. PLoS ONE 5, e9505 (2010).
Kumar, D. K. V. et al. Sci. Transl. Med. 8, 340ra72 (2016).
Eimer, W. A. et al. Neuron 100, 1527–1532 (2018).
Rathore, N. et al. PLoS Genet. 14, e1007427 (2018).
Hur, J.-Y. et al. Nature 586, 735–740 (2020).
 Is ‘friendly fire’ in the brain provoking Alzheimer’s disease?
Is ‘friendly fire’ in the brain provoking Alzheimer’s disease?
 The red hot debate about transmissible Alzheimer’s
The red hot debate about transmissible Alzheimer’s
 How to defeat dementia
How to defeat dementia