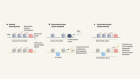
- NEWS AND VIEWS
A mutational signature that can be made by a bacterium arises in human colon cancer
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 580, 194-195 (2020)
doi: https://doi.org/10.1038/d41586-020-00818-7
References
Pleguezuelos-Manzano, C. et al. Nature 580, 269–273 (2020).
Nougayrède, J.-P. et al. Science 313, 848–851 (2006).
Arthur, J. C. et al. Science 338, 120–123 (2012).
Wilson, M. R. et al. Science 363, eaar7785 (2019).
Xue, M. et al. Science 365, eaax2685 (2019).
Priestley, P. et al. Nature 575, 210–216 (2019).
Lee-Six, H. et al. Nature 574, 532–537 (2019).
Olier, M. et al. Gut Microbes 3, 501–509 (2012).
Wassenaar, T. M. Eur. J. Microbiol. Immunol. 6, 147–161 (2016).
Arthur, J. C. et al. Nature Commun. 5, 4724 (2014).
Dejea, C. M. et al. Science 359, 592–597 (2018).
He, Z. et al. Gut 68, 289–300 (2019).
Barrett, M., Hand, C. K., Shanahan, F., Murphy, T. & O’Toole, P. W. Trends Cancer https://doi.org/10.1016/j.trecan.2020.01.019 (2020).
 Read the paper: Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli
Read the paper: Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli
 Bacterial snack attack deactivates a drug
Bacterial snack attack deactivates a drug
 Fungi accelerate pancreatic cancer
Fungi accelerate pancreatic cancer