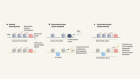
- NEWS AND VIEWS
Brain tumours manipulate neighbouring synapses
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 578, 46-47 (2020)
doi: https://doi.org/10.1038/d41586-020-00137-x
References
Noorani, I. Cancers 11, 1335 (2019).
Allen, N. J. & Lyons, D. A. Science 362, 181–185 (2018).
Yu, K. et al. Nature 578, 166–171 (2020).
Brennan, C. W. et al. Cell 155, 462–477 (2013).
Cancer Genome Atlas Research Network Nature 455, 1061–1068 (2008).
Jung, E. et al. Nature Neurosci. 22, 1951–1960 (2019).
Allen, N. J. et al. Nature 486, 410–414 (2012).
Venkataramani, V. et al. Nature 573, 532–538 (2019).
Venkatesh, H. S. et al. Nature 573, 539–545 (2019).
Barria, A. Nature 573, 499–501 (2019).
Li, N., Gao, W., Zhang, Y.-F. & Ho, M. Trends Cancer 4, 741–754 (2018).
Nishida, T. & Kataoka, H. Cancers 11, 1339 (2019).
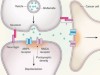
 Read the paper: PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis
Read the paper: PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis
 Drain the swamp to beat glioma
Drain the swamp to beat glioma
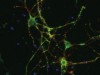
 Dangerous liaisons as tumour cells form synapses with neurons
Dangerous liaisons as tumour cells form synapses with neurons