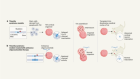
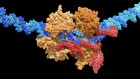
- NEWS FEATURE
Gene therapy is facing its biggest challenge yet

For years, Grajevis Bakatunkanda’s sickle-cell anaemia went undiagnosed. Credit: Aurélie Marrier d'Unienville for Nature
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 576, 22-25 (2019)
doi: https://doi.org/10.1038/d41586-019-03698-8
References
Herrick, J. B. Arch. Intern. Med. 6, 517–521 (1910).
Pauling, L., Itano, H. A., Singer, S. J. & Wells, I. C. Science 110, 543–548 (1949).
Ingram, V. M. Nature 178, 792–794 (1956).
Xu, J. et al. Science 334, 993–996 (2011).
 First CRISPR editing trial results assuage safety concerns
First CRISPR editing trial results assuage safety concerns
 CRISPR deployed to combat sickle-cell anaemia
CRISPR deployed to combat sickle-cell anaemia
 First gene therapy for β-thalassemia approved
First gene therapy for β-thalassemia approved
 Scientists use gene-edited stem cells to treat HIV — with mixed success
Scientists use gene-edited stem cells to treat HIV — with mixed success