- NEWS AND VIEWS
A dynamic view of chemotherapy effectiveness
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 572, 321-322 (2019)
doi: https://doi.org/10.1038/d41586-019-02336-7
References
Campisi, J. & d’Adda di Fagagna, F. Nature Rev. Mol. Cell Biol. 8, 729–740 (2007).
Campisi, J. Annu. Rev. Physiol. 75, 685–705 (2013).
Ewald, J. A., Desotelle, J. A., Wilding, G. & Jarrard, D. F. J. Natl Cancer Inst. 102, 1536–1546 (2010).
Hsu, C.-H., Altschuler, S. J. & Wu, L. F. Cell 178, 361–373 (2019).
d’Adda di Fagagna, F. Nature Rev. Cancer 8, 512–522 (2008).
Fitzgerald, A. L. et al. Cell Death Dis. 6, e1678 (2015).
Abbas, T. & Dutta, A. Nature Rev. Cancer 9, 400–414 (2009).
Potter, A. J. et al. Carcinogenesis 23, 389–401 (2002).
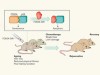 Tools to eliminate senescent cells
Tools to eliminate senescent cells
 Escape from senescence boosts tumour growth
Escape from senescence boosts tumour growth