- NEWS AND VIEWS
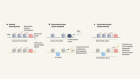
How mutations express themselves in blood-cell production
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 571, 329-330 (2019)
doi: https://doi.org/10.1038/d41586-019-02028-2
References
Nam, A. S. et al. Nature 571, 355–360 (2019).
Kang, H. M. et al. Nature Biotechnol. 36, 89–94 (2018).
Zheng, G. X. Y. et al. Nature Commun. 8, 14049 (2017).
Gupta, I. et al. Nature Biotechnol. 36, 1197–1202 (2018).
Macaulay, I. C. et al. Nature Methods 12, 519–522 (2015).
Buenrostro, J. D. et al. Nature 523, 486–490 (2015).
van Galen, P. et al. Cell 176, 1265-1281 (2019).
 Read the paper: Somatic mutations and cell identity linked by Genotyping of Transcriptomes
Read the paper: Somatic mutations and cell identity linked by Genotyping of Transcriptomes
 Genetic clues can be used to predict whether early-stage cancer will form an invasive tumour
Genetic clues can be used to predict whether early-stage cancer will form an invasive tumour
 Bad blood promotes tumour progression
Bad blood promotes tumour progression