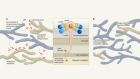
- NEWS AND VIEWS
A licence to kill during inflammation
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 570, 316-317 (2019)
doi: https://doi.org/10.1038/d41586-019-01764-9
References
Sharif, H. et al. Nature 570, 338–343 (2019).
Swanson, K. V., Deng, M. & Ting, J. P.-Y. Nature Rev. Immunol. https://doi.org/10.1038/s41577-019-0165-0 (2019).
Miao, E. A. et al. Nature Immunol. 7, 569–575 (2006).
Franchi, L. et al. Nature Immunol. 7, 576–582 (2006).
Zhang, L. et al. Science 350, 404–409 (2015).
Hu, Z. et al. Science 350, 399–404 (2015).
Schmid-Burgk, J. L. et al. J. Biol. Chem. 291, 103–109 (2016).
Shi, H. et al. Nature Immunol. 17, 250–258 (2016).
He, Y., Zeng, M. Y., Yang, D., Motro, B. & Núñez, G. Nature 530, 354–357 (2016).
Fry, A. M., Bayliss, R. & Roig, J. Front. Cell Dev. Biol. 5, 102 (2017).
Hu, Z. et al. Science 341, 172–175 (2013).
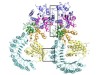 Read the paper: Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome
Read the paper: Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome
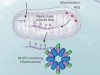 Newly made mitochondrial DNA drives inflammation
Newly made mitochondrial DNA drives inflammation
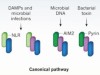 The inflammasome turns 15
The inflammasome turns 15