- NEWS AND VIEWS
A fresh approach to synthesizing ammonia from air and water
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 568, 464-466 (2019)
doi: https://doi.org/10.1038/d41586-019-01213-7
References
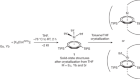
Ashida, Y., Arashiba, K., Nakajima, K. & Nishibayashi, Y. Nature 568, 536–540 (2019).
Hager, T. The Alchemy of Air (Random House, 2009).
Schlögl, R. Angew. Chem. Int. Edn 42, 2004–2008 (2003).
Smil, V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production (MIT Press, 2001).
Appl, M. in Ullmann’s Encyclopedia of Industrial Chemistry 2012 Vol. 3, 139−225 (Wiley, 2012).
Hoffman, B. M., Dean, D. R. & Seefeldt, L. C. Acc. Chem. Res. 42, 609–619 (2009).
Pappas, I. & Chirik, P. J. J. Am. Chem. Soc. 138, 13379−13389 (2016).
Eizawa, A. et al. Nature Commun. 8, 14874 (2017).
Bezdek, M. J., Pappas, I. & Chirik, P. J. Top. Organometal. Chem. 60, 1−21 (2017).
Bezdek, M. J., Guo, S. & Chirik, P. J. Science 354, 730–733 (2016).
Chciuk, T. V. & Flowers, R. A. II J. Am. Chem. Soc. 137, 11526−11531 (2015).
Kolmar, S. S. & Mayer, J. M. J. Am. Chem. Soc. 139, 10687−10692 (2017).
 Read the paper: Molybdenum-catalysed ammonia production with samarium diiodide and alcohols or water
Read the paper: Molybdenum-catalysed ammonia production with samarium diiodide and alcohols or water
 Cyanate fuels the nitrogen cycle
Cyanate fuels the nitrogen cycle
 Methane upgraded by rhodium
Methane upgraded by rhodium