- NEWS AND VIEWS
A mouse model for the most common form of heart failure
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 568, 324-325 (2019)
doi: https://doi.org/10.1038/d41586-019-00983-4
References
Savarese, G. & Lund, L. H. Card. Fail. Rev. 3, 7–11 (2017).
Schiattarella, G. G. et al. Nature 568, 351–356 (2019).
Bloom, M. W. et al. Nature Rev. Dis. Primers 3, 17058 (2017).
Dunlay, S. M., Roger, V. L. & Redfield, M. M. Nature Rev. Cardiol. 14, 591–602 (2017).
Shah, S. J. et al. Circulation 134, 73–90 (2016).
Wang, Z. V. & Hill, J. A. Cell Metab. 21, 215–226 (2015).
Walter, P. & Ron, D. Science 334, 1081–1086 (2011).
Yang, L. et al. Science 349, 500–506 (2015).
Valero-Muñoz, M., Backman, W. & Sam, F. J. Am. Coll. Cardiol. Basic Transl. Sci. 2, 770–789 (2017).
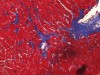 Read the paper: Nitrosative stress drives heart failure with preserved ejection fraction
Read the paper: Nitrosative stress drives heart failure with preserved ejection fraction
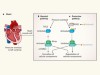 Signalling protein protects the heart muscle from pressure-related stress
Signalling protein protects the heart muscle from pressure-related stress
 Success for pig-to-baboon heart transplants
Success for pig-to-baboon heart transplants