- NEWS AND VIEWS
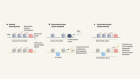
Genetic paradox explained by nonsense
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 568, 179-180 (2019)
doi: https://doi.org/10.1038/d41586-019-00823-5
References
El-Brolosy, M. A. & Stainier, D. Y. R. PLoS Genet. 13, e1006780 (2017).
Kok, F. O. et al. Dev. Cell 32, 97–108 (2015).
Rossi, A. et al. Nature 524, 230–233 (2015).
El-Brolosy, M. A. et al. Nature 568, 193–197 (2019).
Ma, Z. et al. Nature 568, 259–263 (2019).
Zhu, P. et al. J. Genet. Genom. 44, 553–556 (2017).
Popp, M. W.-L. & Maquat, L. E. Annu. Rev. Genet. 47, 139–165 (2013).
Howe, F. S., Fischl, H., Murray, S. C. & Mellor, J. BioEssays 39, 1600095 (2016).
Haimovich. G. et al. Cell 153, 1000–1011 (2013).
Shum, E. Y. et al. Cell 165, 382–395 (2016).
MacArthur, D. G. et al. Science 335, 823–828 (2012).
Brooks, P. C. et al. Cell 79, 1157–1164 (1994).
Bader, B. L., Rayburn, H., Crowley, D. & Hynes, R. O. Cell 95, 507–519 (1998).
 Read the paper: Genetic compensation triggered by mutant mRNA degradation
Read the paper: Genetic compensation triggered by mutant mRNA degradation
 Read the paper: PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components
Read the paper: PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components
 Chromosomes come together to help mice distinguish odours
Chromosomes come together to help mice distinguish odours
 Layered-up regulation in the developing brain
Layered-up regulation in the developing brain