- NEWS AND VIEWS
Structures of STING protein illuminate this key regulator of inflammation
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 567, 321-322 (2019)
doi: https://doi.org/10.1038/d41586-019-00707-8
References
Barber, G. N. Nature Rev. Immunol. 15, 760–770 (2015).
Shang, G. et al. Nature 567, 389–393 (2019).
Zhang, C. et al. Nature 567, 394–398 (2019).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Science 339, 786–791 (2013).
Wu, J. et al. Science 339, 826–830 (2013).
Gall, A. et al. Immunity 36, 120–131 (2012).
Ahn, J., Xia, T., Konno, H., Konno, K., Ruiz, P. & Barber, G. N. Nature Commun. 5, 5166 (2014).
Liu, Y. et al. N. Engl. J. Med. 371, 507–518 (2014).
Sliter, D. A. et al. Nature 561, 258–262 (2018).
Haag, S. M. et al. Nature 559, 269–273 (2018).
Kato, K., Omura, H., Ishitani, R. & Nureki, O. Annu. Rev. Biochem. 86, 541–566 (2017).
Dobbs, N., Burnaevskiy, N., Chen, D., Gonugunta, V. K., Alto, N. M. & Yan, N. Cell Host Microbe. 18, 157–168 (2015).
Liu, S. et al. Science 347, aaa2630 (2015).
Bai, X. C., McMullan, G. & Scheres, S. H. Trends Biochem. Sci. 40, 49–57 (2015).
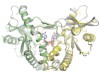 Read the paper: Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP
Read the paper: Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP
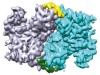 Read the paper: Structural basis of STING binding with and phosphorylation by TBK1
Read the paper: Structural basis of STING binding with and phosphorylation by TBK1
 Genome jail-break triggers lockdown
Genome jail-break triggers lockdown
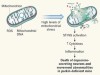 Elusive mitochondrial connection to inflammation uncovered
Elusive mitochondrial connection to inflammation uncovered