
- NEWS AND VIEWS
Bigger is better in virtual drug screens
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 566, 193-194 (2019)
doi: https://doi.org/10.1038/d41586-019-00145-6
References
Bohacek, R. S., McMartin, C. & Guida, W. C. Med. Res. Rev. 16, 3–50 (1996).
Dobson, C. M. Nature 432, 824 (2004).
Dove, A. Nature Biotechnol. 17, 859 (1999).
Lyu, J. et al. Nature 566, 224–229 (2019).
Wootten, D., Christopoulos, A., Marti-Solano, M., Babu, M. M. & Sexton, P. M. Nature Rev. Mol. Cell Biol. 19, 638–653 (2018)
Capuccini, M., Ahmed, L., Schaal, W., Laure, E. & Spjuth, O. J. Cheminformatics 9, 15 (2017).
Ciemny, M. et al. Drug. Discov. Today 23, 1530–1537 (2018).
Kooistra, A. J. et al. Sci. Rep. 6, 28288 (2016).
Renaud, J. P. et al. Nature Rev. Drug Discov. 17, 471–492 (2018).
Rucktooa, P. et al. Sci. Rep. 8, 41 (2018).
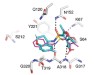 Read the paper: Ultra-large library docking for discovering new chemotypes
Read the paper: Ultra-large library docking for discovering new chemotypes
 Computer model predicts side effects
Computer model predicts side effects
 Designing the ideal opioid
Designing the ideal opioid





