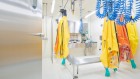
Nick Sireau quit his job to help find an effective treatment for alkaptonuria. Credit: Brian David Stevens
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 565, 148-151 (2019)
doi: https://doi.org/10.1038/d41586-019-00035-x
Updates & Corrections
-
Correction 11 January 2019: An earlier version of this timeline erroneously located the National Alkaptonuria Centre in Cambridge, UK.
References
Garrod, A. E. Lancet 160, 1616–1620 (1902).
Pollak, M. R. et al. Nature Genet. 14, 201–204 (1993).
La Du, B. N., Zannoni, V. G., Laster, L. & Seegmiller, J. E. J. Biol. Chem. 230, 251–260 (1958).
Lindstedt, S., Holme, E., Lock, E. A., Hjalmarson, O. & Strandvik, B. Lancet 340, 813–817 (1992).
Introne, W. J. et al. Mol. Genet. Metab. 103, 307–314 (2011).
Preston, A. J. et al. Ann. Rheum. Dis. 73, 284–289 (2014).
Ranganath, L. R. et al. Mol. Genet. Metab. 125, 127–134 (2018).
 How Facebook and Twitter could be the next disruptive force in clinical trials
How Facebook and Twitter could be the next disruptive force in clinical trials
 Cancer researchers push to relax rules for clinical trials
Cancer researchers push to relax rules for clinical trials
 China embraces precision medicine on a massive scale
China embraces precision medicine on a massive scale
 Personalized medicine: Time for one-person trials
Personalized medicine: Time for one-person trials
 Rare diseases: Genomics, plain and simple
Rare diseases: Genomics, plain and simple