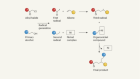
- NEWS AND VIEWS
Practically simple reactions convert hydrocarbons to precious chemicals
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 563, 336-337 (2018)
doi: https://doi.org/10.1038/d41586-018-07333-w
References
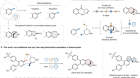
Wang, Z., Yin, H. & Fu, G. C. Nature 563, 379–383 (2018).
Trauner, D. Angew. Chem. Int. Edn 57, 4177–4191 (2018).
Nguyen, L. A., He, H. & Pham-Huy, C. Int. J. Biomed. Sci. 2, 85–100 (2006).
Magano, J. & Dunetz, J. R. Chem. Rev. 111, 2177–2250 (2011).
Lu, X. et al. Nature Commun. 7, 11129–11136 (2016).
Vasseur, A., Bruffaerts, J. & Marek, I. Nature Chem. 8, 209–216 (2016).
Hoveyda, A. H. & Zhugralin, A. R. Nature 450, 243–251 (2007).
Kohler, M. C., Wengryniuk, S. E. & Coltart, D. M. in Stereoselective Synthesis of Drugs and Natural Products (eds Andrushko, V. & Andrushko, N.) 183–213 (Wiley, 2013).
 Read the paper: Catalytic enantioconvergent coupling of secondary and tertiary electrophiles with olefins
Read the paper: Catalytic enantioconvergent coupling of secondary and tertiary electrophiles with olefins
 Reaction combination opens up 3D molecular diversity for drug discovery
Reaction combination opens up 3D molecular diversity for drug discovery
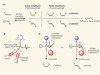 Overcoming catalytic bias
Overcoming catalytic bias