- NEWS AND VIEWS
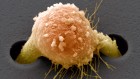
Profile of an unknown airway cell
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 560, 313-314 (2018)
doi: https://doi.org/10.1038/d41586-018-05813-7
References
Montoro, D. T. et al. Nature 560, 319–324 (2018).
Plasschaert, L. W. et al. Nature 560, 377–381 (2018).
Schena, M., Shalon, D., Davis, R. W. & Brown, P. O. Science 270, 467–470 (1995).
Wang, Z., Gerstein, M. & Snyder, M. Nature Rev. Genet. 10, 57–63 (2009).
Tang, F. et al. Nature Methods 6, 377–382 (2009).
Wu, A. R. et al. Nature Methods 11, 41–46 (2014).
Treutlein, B. et al. Nature 509, 371–375 (2014).
Macosko, E. Z. et al. Cell 161, 1202–1214 (2015).
The Tabula Muris Consortium. Preprint at bioRxiv https://dx.doi.org/10.1101/237446 (2017).
Han, X. et al. Cell 172, 1091–1107 (2018).
Howitt, M. R. et al. Science 351, 1329–1333 (2016).
Rock, J. R., Rawlins, E. L., Onaitis, M. W. & Hogan, B. L. Dev. Biol. 331, 503 (2009).
Dymowska, A. K., Hwang, P.-P. & Goss, G. G. Respir. Physiol. Neurobiol. 184, 282–292 (2012).
Elborn, J. S. Lancet 388, 2519–2531 (2016).
Engelhardt, J. F., Zepeda, M., Cohn, J. A., Yankaskas, J. R. & Wilson, J. M. J. Clin. Invest. 93, 737–749 (1994).
 Read the paper: A revised airway epithelial hierarchy includes CFTR-expressing ionocytes
Read the paper: A revised airway epithelial hierarchy includes CFTR-expressing ionocytes
 Read the paper: A single-cell atlas of the airway epithelium reveals the CFTR-rich
Read the paper: A single-cell atlas of the airway epithelium reveals the CFTR-rich
 Bacterial transmission tackled
Bacterial transmission tackled
 Order in the lung
Order in the lung