
- NEWS AND VIEWS
Drug candidate and target for leishmaniasis
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 560, 171-172 (2018)
doi: https://doi.org/10.1038/d41586-018-05765-y
References
Alvar, J. et al. PLoS ONE 7, e35671 (2012).
Wyllie, S. et al. Nature 560, 192–197 (2018).
Vianna, G. Anais do 70 Congresso Brasileiro de Medicina e Cirurgia 4, 426–428 (1912).
Brahmachari, U. A Treatise on Kala Azar (John Bale, Sons & Danielsson, 1928).
World Health Organization. Research priorities for Chagas disease, human African trypanosomiasis and leishmaniasis (WHO, 2012).
Ferguson, F. M. & Gray, N. S. Nature Rev. Drug Discov. 17, 353–357 (2018).
Field, M. C. et al. Nature Rev. Microbiol. 15, 217–231 (2017).
Beneke, T. et al. R. Soc. Open Sci. 4, 170095 (2017).
Jones, N. G., Catta-Preta, C. M. C., Lima, A. P. C. A. & Mottram, J. C. ACS Infect. Dis. 4, 467–477 (2018).
Khare, S. et al. Nature 537, 229–233 (2016).
van Griensven, J. et al. Lancet Infect. Dis. 10, 184–194 (2010).
Rai, K. et al. mBio 4, e00611–13 (2013).
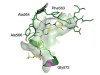 Cyclin-dependent kinase 12 is a drug target for visceral leishmaniasis
Cyclin-dependent kinase 12 is a drug target for visceral leishmaniasis
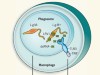 Culprit within a culprit
Culprit within a culprit
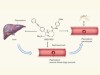 Chemical diversity targets malaria
Chemical diversity targets malaria




